
ceea ce urmează să vă împărtășesc este un ghid pentru clasificarea dispozitivelor medicale.în acest ghid, vă voi oferi o abordare pas cu pas pentru a determina modul în care dispozitivul dvs. medical va fi clasificat de FDA din SUA, Comisia Europeană și Health Canada. Înțelegerea de bază a clasificării normative a produselor va fi de neprețuit pentru eforturile dvs. de a introduce noi produse pe piață.,
clasificare normativă 101
regulile care se aplică dispozitivului dvs. medical depind de modul în care produsul dvs. este clasificat de agențiile de reglementare. Fiecare agenție de reglementare a definit mai multe clasificări diferite pentru dispozitivele medicale.clasificările sunt, în cea mai mare parte sau ca regulă generală, legate de riscul perceput al tipului de produs.
producătorii de dispozitive medicale care vând la nivel internațional trebuie să se familiarizeze cu reglementările aplicabile ale acestor piețe. Acest lucru este mai ușor de zis decât de făcut și poate fi o provocare pentru majoritatea producătorilor., SUA are setul său de reguli, în timp ce Canada aderă la altul, iar Europa încă una.din fericire ,există multe paralele între reglementările și standardele internaționale privind dispozitivele medicale. Acest ghid este conceput pentru a vă arăta cum să clasificați dispozitivul pe diferite piețe din întreaga lume.

de ce clasificarea de reglementare chiar contează?
cunoașterea modului în care dispozitivul dvs. medical este clasificat contează din următoarele motive:
- clasificarea produsului va determina ce trebuie să faceți înainte de a vă putea vinde produsul.,
- clasificarea produsului vă va ajuta să stabiliți cerințe în timpul fazei de dezvoltare a produsului, în special controale de proiectare.
- clasificarea produsului este o componentă importantă în determinarea cât de mult va costa pentru a aduce dispozitivul pe piață și vă va oferi o idee despre cât timp va dura.din acest motiv, vă voi oferi un pic de îndrumare pentru a înțelege mai bine ce să faceți și cum să faceți acest lucru.,notă, informațiile pe care urmează să le furnizez au scopul de a vă ajuta să vă educați cu privire la clasificarea normativă a dispozitivelor medicale și la ceea ce este necesar pentru dispozitivul dvs. medical.
următorul conținut nu este un ghid cuprinzător pentru trimiterile de reglementare, dar ar trebui să vă ofere câteva îndrumări și instrucțiuni de bază privind identificarea modului de stabilire a căii către piață.
o să rămân la „big 3” pe care ar trebui să-l știi când vine vorba de clasificarea dispozitivelor medicale:
- U. S., Alimente & Administrarea Medicamentului, Centrul pentru Dispozitive & Sănătate Radiologică (FDA CDRH)
- Comisia Europeană
- Health Canada
de Reglementare a dispozitivelor Medicale Clasificare în SUA

US FOOD & DRUG ADMINISTRATION (FDA)
În Statele Unite, dispozitivele medicale sunt reglementate de Food & Drug Administration, sau FDA., Ramura specifică din cadrul FDA este Centrul pentru dispozitive & sănătate radiologică (CDRH).misiunea CDRH este de a proteja și promova sănătatea publică. Cu alte cuvinte, asigurați-vă că dispozitivele medicale sunt sigure. În SUA, dispozitivele medicale sunt fie clasa I, Clasa II sau clasa III. clasificarea FDA CDRH se bazează în primul rând pe riscul pe care îl prezintă dispozitivul medical.dispozitivele medicale din clasa I sunt considerate, în general, cu risc scăzut, iar dispozitivele medicale din clasa III sunt considerate ca fiind cel mai mare risc. Tipurile de controale necesare depind de clasificarea produsului dvs.,clasificarea este direct legată de utilizarea prevăzută și de indicațiile de utilizare. Distincția dintre acești termeni este puțin confuză.utilizarea preconizată este scopul general al dispozitivului medical sau funcția acestuia (ceea ce „pretindeți” că face dispozitivul medical).
- indicațiile de utilizare descriu boala sau afecțiunea pe care dispozitivul medical o va diagnostica, trata, preveni, vindeca sau atenua, inclusiv o descriere a populației țintă de pacienți.
țineți cont de acest lucru., Utilizarea prevăzută și indicațiile de utilizare a dispozitivului medical exprimă motivul pentru care ați avut această idee pentru un nou dispozitiv medical.
cum să găsiți reglementările FDA aplicabile pentru dispozitivul dvs. Medical
odată ce definiți utilizarea prevăzută și indicațiile de Utilizare, acum trebuie să găsiți reglementările posibile și codurile produselor. Urmărirea clasificării de reglementare pentru produsul dvs. prin FDA necesită puțin timp și perseverență.,fără a vă plictisi cu prea multe detalii, FDA a stabilit mai multe categorii generale bazate pe specialitatea medicală din CFR titlul 21 – Alimente și medicamente: părți 862 la 892.
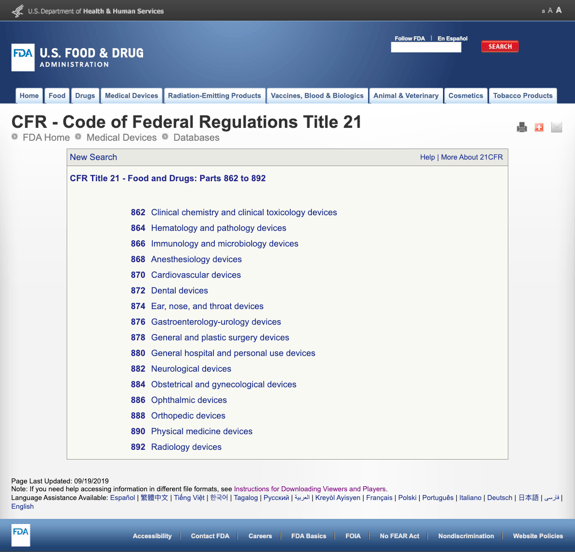
când găsiți categoriile posibile și faceți clic pe numărul regulamentului FDA, lista posibilităților pare brusc nesfârșită. Iată o vedere parțială a opțiunilor pentru dispozitivele cardiovasculare Part 870:

Acest lucru poate fi frustrant și copleșitor.,când găsiți un regulament care pare a fi o posibilă potrivire, puteți să faceți clic pe link și să obțineți mai multe detalii pentru a determina.
De exemplu, dacă eu cred că mi se potrivește de dispozitiv in „870.1250 Percutanata a cateterului,” nu faceți clic pe link-ul și a obține aceste informații:

detaliile furnizate dă-mi o idee dacă utilizarea preconizată și indicațiile de utilizare a alinia cu acest regulament specific. De asemenea, descopăr clasificarea dispozitivelor FDA.,în acest exemplu, aflu că produsul meu este un dispozitiv medical de clasa a II-a(standarde de performanță), ceea ce înseamnă că va trebui să trimit un 510 (k) la FDA înainte de a obține autorizația de piață. Împărtășesc mai multe despre tipurile de observații FDA în continuare în acest ghid.
următorul-găsiți codurile produselor aplicabile dispozitivului dvs.
găsirea Regulamentului aplicabil pentru dispozitivul dvs. medical și clasificarea este prima parte. Acum trebuie să găsiți codurile de produs aplicabile. Iată cum:
Accesați baza de date de clasificare a produselor FDA și introduceți numărul de regulament pe care l-ați găsit., Dacă găsiți mai multe posibilități, atunci va trebui să repetați acest proces pentru fiecare.
când faceți clic pe „Căutare”, veți obține o listă de posibile coduri de produse.
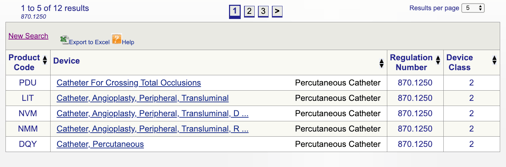
Puteți apoi să examinați fiecare cod individual pentru a determina cea mai bună opțiune pentru produsul dvs. făcând clic pe fiecare cod.
determinarea traseului dvs. către piața din S. U. A.
cunoașterea Regulamentului aplicabil și a codului produsului (așa cum este descris mai sus) este necesară pentru a determina clasificarea dispozitivului dvs. medical., Odată ce aveți aceste informații, veți putea acum să determinați „calea” pentru a vă înregistra produsul la FDA.,
FDA definește trei controale de reglementare pentru fiecare dispozitiv medical clasa:
- Clasa I dispozitiv medical (de la scăzut la risc moderat): Controalele Generale
- Clasa a II-dispozitiv medical (risc moderat până la mare): Controale Generale și Controale Speciale
- Clasa III dispozitive medicale (risc ridicat): Controalele Generale și de Pre-Aprobare (PMA)
lasă-mă Să fiarbă se reduce la asta:
Dacă găsiți produsul dvs. este „scutit”, atunci numai controalele generale se aplică și nu formală FDA este necesară transmiterea. Cu toate acestea, trebuie să vă înregistrați unitatea la FDA și apoi să listați produsul., dacă descoperiți că produsul dvs. necesită controale speciale, aceasta înseamnă că va trebui să pregătiți o depunere de 510(k) către FDA și să primiți clearance-ul înainte de a merge pe piață. După aceea, trebuie să vă înregistrați unitatea și să listați produsul.dacă găsiți că produsul dvs. necesită aprobare premarket, aceasta înseamnă că va trebui să urmați procesul FDA PMA pentru a primi aprobarea înainte de a merge pe piață.,
Dispozitiv Medical de Clasificare în Europa

Comisia Europeană
regulamentului pentru un dispozitiv medical în Uniunea Europeană (UE) sunt stabilite prin Directivele privind dispozitivele Medicale de către Comisia Europeană (CE).calea către piața din Europa este obținerea unui marcaj CE.pentru a afla ce este necesar pentru a obține marcajul CE al dispozitivului dumneavoastră medical, trebuie mai întâi să stabiliți clasificarea UE a dispozitivului dumneavoastră medical., Regulamentul Uniunii Europene privind dispozitivele medicale (eu MDR) include informațiile necesare pentru a determina clasa dispozitivului dvs.
UE MDR 2017/745 modifică Directiva 2001/83/CE, Regulamentul (CE) Nr. 178/2002 și Regulamentul (CE) Nr 1223/2009 și de abrogare a Directivelor 90/385/CEE și 93/42/CEE. MDR din UE va deveni regulamentul obligatoriu pentru dispozitivele medicale începând cu luna mai 2020.va trebui să stabiliți dacă dispozitivul dumneavoastră medical este:
- Non-invaziv
- orice dispozitiv care nu penetrează corpul printr-un orificiu sau prin suprafața corpului., Aceste dispozitive sunt de obicei clasa I; cu toate acestea, se aplică anumite reguli și excepții care le-ar putea face clasa II sau mai mare. orice dispozitiv care, integral sau parțial, pătrunde în interiorul corpului, fie printr-un orificiu al corpului, fie prin suprafața corpului.orice dispozitiv a cărui funcționare depinde de o altă sursă de energie decât cea generată de corpul uman în acest scop sau de gravitație și care acționează prin schimbarea densității sau transformarea acelei energii.,pentru fiecare dintre categoriile mari, există anumite reguli care se aplică, prezentate în anexa VIII a noului regulament privind dispozitivele medicale. Aceste categorii, împreună cu durata de utilizare, fac ca determinarea Clasificării să fie destul de simplă. de exemplu, un dispozitiv în utilizare continuă timp de sub 60 de minute este considerat o durată tranzitorie, 60 de minute până la 30 de zile este considerat pe termen scurt, iar peste 30 de zile este considerat pe termen lung.,având în vedere acest lucru, pentru a determina clasificarea UE a dispozitivului dvs., putem folosi exemplul cateterului percutanat utilizat anterior în acest ghid pentru clasificarea FDA.să presupunem că stabilesc că dispozitivul meu medical se încadrează în categoria „invazivă”; acest lucru restrânge căutarea mea până la regulile 5, 6, 7 și 8.
pot restrânge Regulile în continuare, deoarece știu că dispozitivul meu medical este pe termen scurt, deoarece este utilizat pentru o perioadă mai mare de 24 de ore și mai puțin de 30 de zile.de acolo, pot determina că regula 7 este cea mai aplicabilă clasificării dispozitivului meu.,

Sursa: UE MDR 2017/ 745 Anexa VIII 5.3
Când mi-percutanata a cateterului va administra medicamente, pot să confirm că mi dispozitiv medical este considerat o Clasă IIb dispozitiv medical în marea piață Europeană.
determinarea traseului dvs. către piața din Europa
Uniunea Europeană are un sistem de clasificare a produselor similar cu cel din SUA.,:
- clasa I
- clasa IIa
- clasa IIb
- clasa III
în toate cazurile pentru dispozitivele medicale care urmează să fie vândute în Uniunea Europeană, documentația tehnică este o etapă necesară în procesul de obținere a marcajului CE. Toate clasele de dispozitive medicale din UE necesită colaborarea cu un organism notificat, cu excepția celor care sunt din clasa I și care pot fi autocertificate.companiile vor trebui, de asemenea, să lucreze cu un reprezentant autorizat pentru a avea grijă de înregistrarea produselor în Europa., Articolul 11 Cele mai importante întrebări cu privire la reprezentanții autorizați ai ce pot contribui la clarificarea suplimentară a modului de realizare a clasificării dispozitivelor medicale în Europa.
clasificarea dispozitivelor medicale în Canada

Health Canada
reglementările privind dispozitivele medicale din Canada sunt stabilite de Guvernul Canadei și reglementate de Health Canada.la fel ca SUA și UE, pentru a vinde pe piața canadiană, trebuie să stabiliți mai întâi clasificarea dispozitivelor medicale în conformitate cu regulamentul Canadei.,Similar cu cerințele prezentate în MDR din UE, Health Canada oferă o îndrumare destul de simplă și ușor de urmat privind Sistemul de clasificare bazat pe risc pentru dispozitivele de Diagnostic Non-in Vitro pe care producătorii de dispozitive medicale le pot utiliza atunci când vând pe această piață.,
Health Canada definește patru grupuri de non-medicale pentru diagnostic in vitro dispozitive:
- Dispozitive Invazive (Regulile 1 – 3)
- Dispozitive Non-Invazive (Articolele 4 – 7)
- Dispozitive Active (Regulile 8 – 12)
- Reguli Speciale (Articolele 13 – 16)
Pentru fiecare dintre categorii, există un set de reguli care se aplică. Aceste reguli sunt ceea ce producătorii ar trebui să urmeze pentru a determina clasificarea riscurilor dispozitivului lor.
voi folosi exemplul cateterului percutanat încă o dată în cazul comercializării unui astfel de dispozitiv în Canada.,
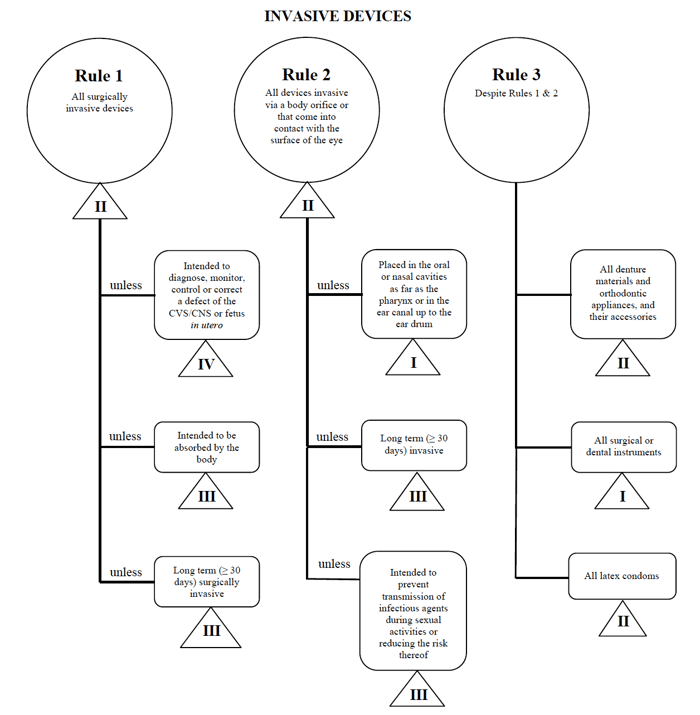
Sursa: Orientări privind Riscul bazat pe Sistemul de Clasificare pentru Non-Diagnostic In Vitro
nu-mi determine dispozitiv medical se încadrează în „invazive” categorie, îngustarea căutarea mea până la Regulile 1, 2, și 3.
după examinarea opțiunilor, stabilesc că se aplică regula 1.pe baza destinației mele, dispozitivul meu medical este considerat clasa a II-a în Canada.,există patru niveluri de clasificări ale dispozitivelor medicale în Canada:
- clasa I
- clasa II
- clasa III
- clasa IV
înainte de a intra pe piață în Canada, trebuie să solicitați mai întâi o licență pentru dispozitive medicale. Dispozitivele medicale din clasa I nu necesită licență. Producătorii pot face referire la documentul de orientare Health Canada, care vă ghidează prin acest proces.,
producătorii de dispozitive medicale din clasa III și clasa IV pot primi licența prin trimiterea unei cereri premarket, în formatele ToC sau Health Canada, pentru intrarea pe piața canadiană.de asemenea, va trebui să obțineți certificarea ISO 13485 cu MDSAP.din ianuarie 2019, toți producătorii de dispozitive medicale care vând dispozitive medicale de clasa II și mai mari pe piața canadiană trebuie să facă parte din programul de Audit unic al dispozitivelor medicale (MDSAP).,
acești producători trebuie să fie supuși și să treacă un audit complet al sistemului lor de management al calității (SMC) prin programul din Canada. În prezent, există 6 regiuni din întreaga lume care participă la MDSAP, inclusiv Canada, SUA, Japonia, Brazilia și Australia.în afară de acei producători solicitați de Health Canada să participe la program, participarea la MDSAP este opțională pentru producători.,
Pentru a obține MDSAP de certificare, producătorii de dispozitive medicale trebuie să completeze următoarele trei etape:
- Aplicarea și revizuirea
- Off-ului documentația de audit
- Pe site-ul de audit
factorii de decizie Dispozitiv de vânzare în Canada vor fi supuse unei revizuiri anuale, cu un audit de recertificare la fiecare al treilea an. Conformitatea cu programul de Audit unic al dispozitivelor medicale se bazează pe respectarea ghidurilor din standardul ISO 13485 privind sistemele de management al calității pentru dispozitivele medicale.,vă recomandăm instruirea echipelor dvs. de produse și de calitate și de reglementare cu privire la cerințele MDSAP aplicabile pentru a vă eficientiza aplicația.
Nostru gratuit decalaj instrument de evaluare pentru MDSAP și ISO 13485 ajută factorii de decizie dispozitiv evaluarea SMC recomandările din standardul ISO alături de cerințele de Sănătate Canada audit organizații (AO) și alte regiuni participante la program.,
ultimele Gânduri
acum Aveți informațiile și resursele de care aveți nevoie pentru a determina dispozitiv medical de clasificare și de calea sa de piață în trei dintre cele mai mari piețe din întreaga lume. indiferent de clasificarea dispozitivului, este imperativ să respectați regulile de reglementare care se aplică pe fiecare piață. Respectarea conformității este un aspect cheie al managementului calității, care va decide în cele din urmă soarta dispozitivului și a companiei în ansamblu.,la Greenlight Guru, apreciem importanța managementului calității dispozitivelor medicale (MDQMS). Software-ul nostru MDQMS este singura soluție din lume concepută special pentru dispozitivele medicale. Este aliniat cu cele mai recente practici standard din domeniu pentru gestionarea controalelor de proiectare a produselor și a riscurilor, precum și a activităților de control al schimbărilor și a altor evenimente de calitate, oferind o trasabilitate completă pe tot parcursul ciclului de viață al dispozitivului dumneavoastră medical.
căutați o soluție de control al proiectării care să vă ajute să aduceți dispozitive medicale mai sigure pe piață mai rapid, cu un risc mai mic?, Faceți clic aici pentru a face un tur rapid al software-ului SMC al dispozitivului medical Greenlight Guru →