
Hvad jeg er ved at dele med dig er en guide til medicinsk enhed forskriftsmæssige klassificering.
i denne vejledning vil jeg give dig en trinvis tilgang til at bestemme, hvordan dit medicinske udstyr vil blive klassificeret af US FDA, Europa-Kommissionen og Health Canada. At få en grundlæggende forståelse af lovgivningsmæssig produktklassificering vil være uvurderlig for din indsats for at bringe nye produkter på markedet.,
Reguleringsklassificering 101
de regler, der gælder for dit medicinske udstyr, afhænger af, hvordan dit produkt klassificeres af reguleringsagenturerne. Hvert reguleringsagentur har defineret flere forskellige klassifikationer for medicinsk udstyr.
klassificeringerne er for det meste eller som hovedregel relateret til den opfattede risiko ved produkttypen.producenter af medicinsk udstyr, der sælger internationalt, skal gøre sig bekendt med de gældende regler på disse markeder. Dette er lettere sagt end gjort og kan være en udfordring for de fleste producenter., USA har sit sæt regler, mens Canada overholder en anden, og Europa en anden stadig.
heldigvis er der mange paralleller mellem internationale forskrifter for medicinsk udstyr og standarder. Denne vejledning er designet til at vise dig, hvordan du klassificerer din enhed på forskellige markeder rundt om i verden.

hvorfor betyder Reguleringsklassificering endda noget?
at vide, hvordan dit medicinske udstyr er klassificeret, betyder noget af følgende årsager:
- produktklassificering bestemmer, hvad du skal gøre, før du kan sælge dit produkt.,
- produktklassificering hjælper dig med at etablere krav i produktudviklingsfasen, specifikt designkontroller.produktklassificering er en vigtig komponent til at bestemme, hvor meget det vil koste at bringe din enhed på markedet og give dig en ID.om, hvor lang tid det vil tage.
På grund af dette vil jeg give dig en lille smule vejledning for bedre at forstå, hvad du skal gøre, og hvordan du gør det.,
Bemærk, de oplysninger, jeg er ved at give, er beregnet til at hjælpe dig med at uddanne dig om klassificering af medicinsk udstyr, og hvad der kræves for dit medicinske udstyr.
følgende indhold er ikke en omfattende guide til regulatoriske indlæg, men bør give dig nogle grundlæggende vejledning og retning om at identificere, hvordan man etablerer path to market.
Jeg holder mig til den “store 3”, Du skal vide, når det kommer til klassificering af medicinsk udstyr:
- U. S., Mad & Drug Administration, Center for Enheder & Radiological Health (FDA CDRH)
- Europa-Kommissionen
- Health Canada
Medicinsk udstyr Forskriftsmæssige Klassificering i USA

U.S. FOOD & DRUG ADMINISTRATION (FDA)
I de Forenede Stater, medicinsk udstyr er reguleret af Food & Drug Administration, FDA., Den specifikke gren inden for FDA er Center for enheder & Radiologisk Sundhed (CDRH).
CDRH ‘ s mission er at beskytte og fremme folkesundheden. Med andre ord, sørg for, at medicinsk udstyr er sikkert. I USA er medicinsk udstyr enten klasse i, klasse II eller klasse III. FDA CDRH-klassificeringen er primært baseret på risiko, som det medicinske udstyr udgør.
medicinsk udstyr i klasse I anses generelt for lav risiko, og medicinsk udstyr i klasse III betragtes som den højeste risiko. De typer kontroller, der kræves, afhænger af dit produkts klassificering.,
klassificering er direkte relateret til beregnet brug og indikationer for brug. Sondringen mellem disse udtryk er lidt forvirrende.
- Beregnet anvendelse er det generelle formål med det medicinske udstyr eller dets funktion (hvad du “hævder” det medicinske udstyr gør).
- indikationer for anvendelse beskriv den sygdom eller tilstand, som det medicinske udstyr vil diagnosticere, behandle, forebygge, helbrede eller afbøde, herunder en beskrivelse af målpatientpopulationen.
husk dette., Den påtænkte anvendelse og indikationer for brug af dit medicinske udstyr udtrykker grunden til, at du havde denne ID.til et nyt medicinsk udstyr.
Sådan finder du de gældende FDA-regler for dit medicinske udstyr
Når du har defineret Tilsigtet brug og indikationer for brug, skal du nu finde de mulige regler og produktkoder. Sporing af reguleringsklassificering for dit produkt via FDA tager lidt tid og udholdenhed.,
uden at kede dig med for mange detaljer, har FDA etableret flere generelle kategorier baseret på den medicinske specialitet i CFR Titel 21 – mad og medicin: dele 862 til 892.
Når du finder de mulige kategorier og klikker på FDA-reguleringsnummeret, synes listen over muligheder pludselig uendelig. Her er en delvis visning af mulighederne for Del 870 kardiovaskulære enheder:

dette kan være frustrerende og overvældende.,
Når du finder en regulering, der ser ud til at være en mulig pasform, kan du klikke på linket og få flere detaljer for at bestemme.
For eksempel, hvis jeg tror, min enhed passer i “870.1250 Perkutan kateter,” jeg klikker på linket og få denne information:

De oplysninger, der leveres give mig en ide, om min påtænkte brug og indikationer for brug justere med denne særlige regulering. Jeg opdager også FDA-enhedsklassifikationen.,
i dette eksempel lærer jeg, at mit produkt er en klasse II medicinsk udstyr (præstationsstandarder), hvilket betyder, at jeg bliver nødt til at indsende en 510(k) til FDA, før jeg får markedsgodkendelse. Jeg deler mere om typer af FDA indlæg videre i denne vejledning.
næste – Find de produktkoder, der gælder for din enhed
find den gældende forordning for dig medicinsk udstyr og klassificering er den første del. Nu skal du finde de gældende produktkoder. Sådan gør du:
gå til FDA-Produktklassifikationsdatabasen, og skriv det reguleringsnummer, du fandt., Hvis du finder mere end en mulighed, skal du gentage denne proces for hver.
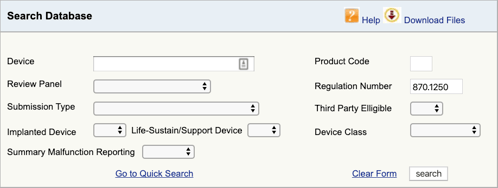
Når du klikker på “Søg”, får du en liste over mulige produktkoder.
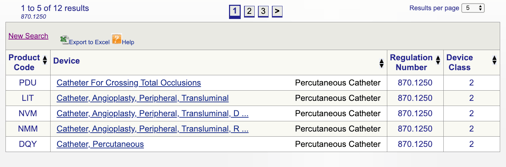
Du kan derefter gennemgå hver enkelt kode for at bestemme den bedste mulighed for dit produkt ved at klikke på hver kode.
det er nødvendigt for dig at bestemme din vej til markedet i USA
at kende den gældende regulering og Produktkode (som beskrevet ovenfor) for at bestemme klassificeringen af dit medicinske udstyr., Når du har disse oplysninger, vil du nu kunne bestemme “stien” for at få dit produkt registreret hos FDA.,
FDA definerer tre regulerende kontrol for hver medicinsk udstyr klasse:
- Klasse i medicinsk udstyr (lav til moderat risiko): Generel Kontrol
- Class II medical device (moderat til høj risiko): Generel Kontrol og Særlige Kontroller
- Class III medicinsk udstyr (høj risiko): Generel Kontrol og Godkendelse før markedsføring (PMA)
Lad mig koge det ned til dette:
Hvis du kan finde dit produkt, er “fritaget”, så kun generel kontrol, og ingen formelle FDA indsendelse er påkrævet. Du behøver dog at registrere din virksomhed med FDA og derefter Liste produktet.,
Hvis du finder, at dit produkt kræver særlige kontroller, betyder det, at du bliver nødt til at forberede en 510(k) indsendelse til FDA og modtage godkendelse, før du går på markedet. Derefter skal du registrere din virksomhed og liste produktet.
Hvis du finder ud af, at dit produkt kræver forhåndsgodkendelse, betyder det, at du bliver nødt til at følge FDA PMA-processen for at modtage godkendelse, før du går på markedet.,
Medicinsk udstyr Klassificering i Europa

Europa-Kommissionen
reglerne for medicinsk udstyr i den Europæiske Union (EU) er etableret gennem Direktiverne om Medicinsk udstyr, som Europa-Kommissionen (EF).
vejen til markedet i Europa er at opnå en CE-mærkning.
for at finde ud af, hvad der kræves for at få en CE-mærkning af dit medicinske udstyr, skal du først bestemme EU-klassificeringen af dit medicinske udstyr., Den Europæiske Unions forordning om medicinsk udstyr (EU MDR) indeholder de nødvendige oplysninger til at bestemme din enhedsklasse.
EU MDR 2017/745 ændring af Direktiv 2001/83/EF, Forordning (EF) 178/2002 og Forordning (EF) Nr 1223/2009 og om ophævelse af Rådets Direktiv 90/385/EØF og 93/42/EØF. EU MDR bliver den obligatoriske regulering for medicinsk udstyr, der starter i maj 2020.
Du skal afgøre, om dit medicinske udstyr er:
- ikke-invasiv
- ethvert udstyr, der ikke trænger ind i kroppen gennem en åbning eller overfladen af kroppen., Disse enheder er typisk klasse I; dog gælder visse regler og undtagelser, der kan gøre dem til klasse II eller højere.
- invasiv
- enhver anordning, som helt eller delvist trænger ind i kroppen, enten gennem en kropsåbning eller gennem overfladen af kroppen.
- Aktiv
- enhver enhed, hvis funktion afhænger af en anden energikilde end den, der genereres af den menneskelige krop til dette formål, eller af tyngdekraften, og som virker ved at ændre densiteten af eller konvertere den energi.,
for hver af de brede kategorier er der visse regler, der gælder, som beskrevet i bilag VIII til den nye forordning om medicinsk udstyr. Disse kategorier kombineret med varigheden for brug gør afgørende klassificering ret ligetil. for eksempel betragtes en enhed i kontinuerlig brug i Under 60 minutter som kortvarig varighed, 60 minutter til 30 dage betragtes som kortvarig, og over 30 dage betragtes som langvarig.,
med det i tankerne, for at bestemme EU-klassificeringen af din enhed, kan vi bruge det perkutane katetereksempel, der blev brugt tidligere i denne vejledning til FDA-klassificering.
lad os sige, at jeg bestemmer, at min medicinske enhed passer ind i kategorien “invasiv”; dette indsnævrer min søgning ned til Regler 5, 6, 7 og 8.
Jeg kan derefter indsnævre reglerne yderligere, da jeg ved, at min medicinske enhed er kortvarig, fordi den bruges i en periode større end 24 timer og mindre end 30 dage.
derfra er jeg i stand til at bestemme, at Regel 7 gælder mest for klassificeringen af min enhed.,

Kilde: EU MDR 2017/ 745 Bilag VIII 5.3
Da min perkutan kateter vil administrere lægemidler, kan jeg bekræfte, at min medicinske udstyr anses for en Klasse IIb, medicinsk enhed i det Europæiske marked.
bestemmelse af din vej til markedet i Europa
Den Europæiske Union har et lignende produktklassificeringssystem som USA,:
- klasse I
- klasse IIa
- klasse IIb
- klasse III
i alle tilfælde for medicinsk udstyr, der skal sælges i Den Europæiske Union, er teknisk dokumentation et påkrævet trin i processen med at opnå CE-mærkning. Alle klasser af medicinsk udstyr i EU kræver samarbejde med et bemyndiget organ, bortset fra dem, der er klasse I og kan være selvcertificerede.
virksomheder skal også arbejde med en autoriseret repræsentant for at tage sig af produktregistrering i Europa., Artikel 11 de vigtigste spørgsmål om EF-bemyndigede repræsentanter kan bidrage til yderligere afklaring af, hvordan man opnår klassificering af medicinsk udstyr i Europa.
Medicinsk udstyr Klassificering i Canada
![]()
Health Canada
medicinsk udstyr bestemmelser i Canada er etableret af den canadiske Regering og er reguleret af Health Canada.
ligesom USA og EU skal du først bestemme klassificeringen af medicinsk udstyr i henhold til Canadas regulering for at sælge til den canadiske markedsplads.,
i lighed med kravene i EU MDR giver Health Canada en ret enkel og let at følge vejledning om det risikobaserede klassifikationssystem for ikke-in Vitro-diagnostiske udstyr, som producenter af medicinsk udstyr kan bruge, når de sælger til dette marked.,
Health Canada definerer fire grupper af ikke-in-vitro-diagnostisk medicinsk udstyr:
- Invasive Enheder (Regler 1 – 3)
- Ikke-Invasivt Udstyr (Regler 4 – 7)
- Aktive Enheder (regel 8 – 12)
- Særlige Regler (regel 13 – 16)
For hver af de overordnede kategorier, der er et sæt regler, der gælder. Disse regler er, hvad producenterne skal følge for at bestemme risikoklassificeringen af deres enhed.
Jeg bruger det perkutane katetereksempel endnu en gang i tilfælde af markedsføring af en sådan enhed i Canada.,
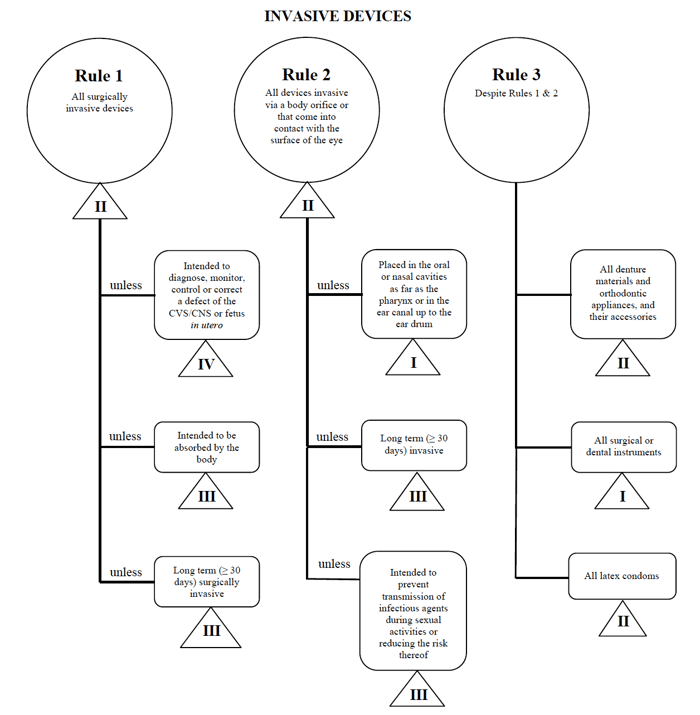
kilde: vejledning i det risikobaserede klassificeringssystem for ikke-in Vitro-diagnostiske udstyr
Jeg bestemmer, at mit medicinske udstyr passer ind i kategorien “invasiv” og begrænser min søgning til Regel 1, 2 og 3.
efter at have gennemgået indstillingerne bestemmer jeg, at Regel 1 gælder.
baseret på min tilsigtede anvendelse betragtes mit medicinske udstyr som klasse II i Canada.,
FASTSÆTTELSEN af DIN VEJ TIL MARKEDET I CANADA
Der er fire niveauer af medicinsk udstyr klassifikationer i Canada:
- Klasse jeg
- i Klasse II
- Class III
- i Klasse IV
Forud for at gå til marked i Canada, skal du først ansøge om et medical device-licens. Klasse i medicinsk udstyr kræver ikke en licens. Producenter kan henvise til Health Canada guidance document, som leder dig gennem denne proces.,
Fabrikanter af Klasse III og Klasse IV, medicinsk udstyr, der kan modtage deres licens ved at indsende en før markedsføring ansøgning, enten i Indholdsfortegnelsen eller Health Canada formater, for at komme ind på det Canadiske marked.
Du skal også få ISO 13485-certificering med mdsap.
opdatering til Health Canada Regulations: mdsap
fra januar 2019 skal alle producenter af medicinsk udstyr, der sælger klasse II medicinsk udstyr og højere til det canadiske marked, være en del af Medical Device Single Audit Programme (mdsap).,
Disse producenter skal gennemgå og bestå en fuld revision af deres kvalitetsstyringssystem (QMS) gennem programmet i Canada. Der er i øjeblikket 6 regioner rundt om i verden, der deltager i mdsap, herunder Canada, USA, Japan, Brasilien og Australien.
bortset fra de producenter, der kræves af Health Canada for at deltage i programmet, er deltagelse i mdsap valgfri for producenterne.,
til At få MDSAP certificering, producenter af medicinsk udstyr, skal du udføre følgende tre trin:
- Ansøgning og anmeldelse
- Off-site dokumentation revision
- On-site audit
Enhed-producenter sælger i Canada vil blive udsat for årlige anmeldelser, med en recertificering revision hvert tredje år. Overholdelse af Single Audit-programmet for medicinsk udstyr er baseret på at opfylde retningslinjerne fra ISO 13485-standarden om kvalitetsstyringssystemer for medicinsk udstyr.,
Vi anbefaler at træne dit produkt-og kvalitets-og reguleringshold i de gældende MDSAP-krav for at strømline din ansøgning.
Vores gratis hul assessment tool for MDSAP og ISO 13485 hjælper enheden beslutningstagere vurderer KVALITETSSTYRINGSSYSTEMET retningslinjer fra ISO-standarden sammen med kravene til Health Canada revision organisationer (AO) og andre regioner, der deltager i programmet.,
Sidste Tanker
Du har nu de oplysninger og ressourcer, er du nødt til at bestemme din medicinske enhed, for klassificering og vej til markedet i tre af de største markedspladser rundt om i verden.
uanset klassificeringen af din enhed er det bydende nødvendigt, at du følger de lovgivningsmæssige retningslinjer, der gælder på hvert marked. Overholdelse af overholdelse er et centralt aspekt af kvalitetsstyring, der i sidste ende bestemmer skæbnen for din enhed og virksomhed som helhed.,
på Greenlight Guru værdsætter vi vigtigheden af medicinsk udstyrskvalitetsstyring (MD .ms). Vores MD .ms-soft .are er den eneste løsning i verden designet specielt til medicinsk udstyr. Det er i overensstemmelse med den nyeste branchestandard bedste praksis for styring af produktdesign kontrol og risiko, samt ændre kontrolaktiviteter og andre kvalitetsbegivenheder, hvilket giver fuld sporbarhed i hele livscyklussen for dit medicinske udstyr.
Leder du efter en designstyringsløsning, der hjælper dig med at bringe sikrere medicinsk udstyr på markedet hurtigere med mindre risiko?, Klik her for at tage en hurtig rundvisning i Greenlight Guru Medical Device Medicalms soft /are