
ce que je vais partager avec vous est un guide de classification réglementaire des instruments médicaux.
dans ce guide, je vais vous fournir une approche étape par étape pour déterminer comment votre instrument médical sera classé par la FDA des États-Unis, La Commission européenne et Santé Canada. Obtenir une compréhension de base de la classification réglementaire des produits sera inestimable pour vos efforts pour mettre de nouveaux produits sur le marché.,
Classification Réglementaire 101
Les règles qui s’appliquent à votre appareil médical dépend de la façon dont votre produit est classé par les organismes de réglementation. Chaque organisme de réglementation a défini plusieurs classifications différentes pour les dispositifs médicaux.
Les classifications sont, pour la plupart ou en règle générale, liées au risque perçu du type de produit.
Les fabricants de dispositifs médicaux qui vendent à l’échelle internationale doivent se familiariser avec la réglementation applicable de ces marchés. C’est plus facile à dire qu’à faire et cela peut être un défi pour la plupart des fabricants., Les États-Unis ont leur ensemble de règles, tandis que le Canada adhère à une autre, et L’Europe une autre encore.
heureusement, il existe de nombreux parallèles entre les réglementations et les normes internationales relatives aux dispositifs médicaux. Ce guide est conçu pour vous montrer comment classer votre appareil sur différents marchés à travers le monde.

pourquoi la Classification réglementaire est-elle même importante?
savoir comment votre instrument médical est classé est important pour les raisons suivantes:
- La classification du produit déterminera ce que vous devez faire avant de pouvoir vendre votre produit.,
- La classification des produits vous aidera à établir les exigences pendant la phase de développement du produit, en particulier les contrôles de conception.
- La classification des produits est un élément important pour déterminer combien il en coûtera pour mettre votre appareil sur le marché et vous donner une idée du temps que cela prendra.
de ce fait, je vais vous fournir avec un peu de conseils pour mieux comprendre ce qu’il faut faire et comment le faire.,
Remarque, les renseignements que je vais vous fournir visent à vous renseigner sur la classification réglementaire des instruments médicaux et sur ce qui est requis pour votre instrument médical.
le contenu suivant n’est pas un guide complet pour les présentations réglementaires, mais devrait vous donner des conseils et des directives de base sur la façon d’établir la voie d’accès au marché.
je vais m’en tenir au « big 3 » que vous devriez connaître en matière de classification des dispositifs médicaux:
- U. S., Food & Drug Administration, Center for Devices & Radiological Health (FDA CDRH)
- Commission européenne
- Santé Canada
Medical Device Regulatory Classification in the U. S.

U. S. Food & Drug Administration (FDA)
aux États-Unis, les dispositifs médicaux sont réglementés par la Food & Drug Administration, ou FDA., La branche spécifique au sein de la FDA est le Center for Devices & Radiological Health (CDRH).
la mission du CDRH est de protéger et de promouvoir la santé publique. En d’autres termes, assurez-vous que les dispositifs médicaux sont sûrs. Aux États-Unis, les instruments médicaux sont de classe I, de classe II ou de classe III. la classification CDRH de la FDA est basée principalement sur le risque que pose le dispositif médical.
Les instruments médicaux de classe I sont généralement considérés comme présentant un faible risque et les instruments médicaux de classe III comme présentant le risque le plus élevé. Les types de contrôles requis dépendent de la classification de votre produit.,
la Classification est directement liée à l’utilisation prévue et aux indications d’utilisation. La distinction entre ces termes est un peu déroutante.
- l’utilisation prévue est l’usage général du dispositif médical ou de sa fonction (ce que vous « revendiquez” du dispositif médical).
- Indications d’utilisation décrire la maladie ou la condition que l’instrument médical diagnostiquera, traitera, préviendra, guérira ou atténuera, y compris une description de la population de patients cible.
Gardez cela à l’esprit., L’utilisation prévue et les indications d’utilisation de votre dispositif médical expriment la raison pour laquelle vous avez eu cette idée d’un nouveau dispositif médical.
Comment trouver les réglementations de la FDA applicables à votre dispositif médical
Une fois que vous avez défini l’utilisation prévue et les indications d’utilisation, vous devez maintenant trouver les réglementations et les codes de produit possibles. Le suivi de la classification réglementaire de votre produit via la FDA prend un peu de temps et de persévérance.,
sans vous ennuyer avec trop de détails, la FDA a établi plusieurs catégories générales basées sur la spécialité médicale dans CFR titre 21 – aliments et médicaments: parties 862 à 892.
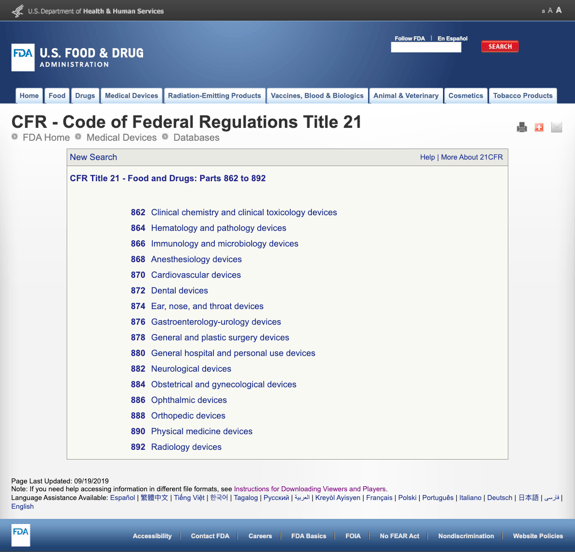
lorsque vous trouvez les catégories possibles et cliquez sur le numéro de règlement FDA, la liste des possibilités semble soudainement infinie. Voici une vue partielle des options pour la Partie 870 appareils Cardiovasculaires:

Cela peut être frustrant et écrasante.,
Lorsque vous trouvez un règlement, qui semble être un ajustement possible, vous pouvez cliquer sur le lien et obtenir plus de détails à prendre une décision.
par exemple, si je pense que mon appareil correspond au cathéter percutané « 870.1250”, je clique sur le lien et j’obtiens cette information:

Les détails fournis me donnent une idée si mon utilisation prévue et les indications d’utilisation sont conformes à cette réglementation spécifique. Je découvre également la classification des appareils FDA.,
dans cet exemple, j’apprends que mon produit est un dispositif médical de classe II (normes de performance), ce qui signifie que je devrai soumettre un 510(k) À LA FDA avant d’obtenir l’autorisation de mise sur le marché. Je partage plus sur les types de soumissions FDA plus loin dans ce guide.
suivant – trouver les Codes de produit applicables à votre appareil
trouver la réglementation applicable pour vous Dispositif médical et la classification est la première partie. Vous devez maintenant trouver les codes de produit applicables. Voici comment:
accédez à la base de données de Classification des produits de la FDA et tapez le numéro de règlement que vous avez trouvé., Si vous trouvez plus d’une possibilité, vous devrez répéter ce processus pour chacun.
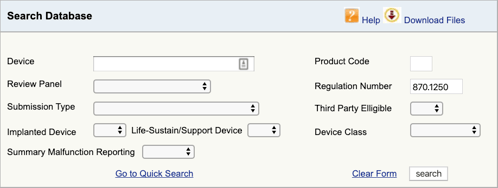
Lorsque vous cliquez sur « rechercher”, vous obtiendrez une liste de codes de produit.
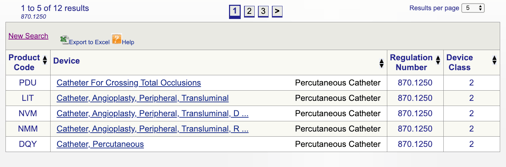
Vous pouvez ensuite passer en revue chaque code à déterminer la meilleure option pour votre produit en cliquant sur chaque code.
déterminer votre chemin vers le marché aux États-Unis
Il est nécessaire de connaître la réglementation applicable et le code du produit (tel que décrit ci-dessus) pour déterminer la classification de votre instrument médical., Une fois que vous avez ces informations, vous serez maintenant en mesure de déterminer le « chemin” pour obtenir votre produit enregistré auprès de la FDA.,
la FDA définit trois contrôles réglementaires pour chaque classe de dispositifs médicaux:
- dispositifs médicaux de classe I (risque faible à modéré): contrôles généraux
- dispositifs médicaux de classe II (risque modéré à élevé): contrôles généraux et contrôles spéciaux
- dispositifs médicaux de classe III (risque élevé): contrôles généraux et approbation avant commercialisation (PMA)
laissez-moi résumer à ceci:
Si vous ensuite, seuls des contrôles généraux s’appliquent et aucune soumission formelle à la FDA n’est requise. Cependant, vous devez enregistrer votre établissement auprès de la FDA, puis lister le produit.,
Si vous trouvez que votre produit nécessite des contrôles spéciaux, cela signifie que vous devrez préparer une soumission 510(k) À LA FDA et recevoir une autorisation avant d’aller sur le marché. Après cela, vous devez enregistrer votre établissement et lister le produit.
Si vous trouvez que votre produit nécessite une approbation avant la mise sur le marché, cela signifie que vous devrez suivre le processus PMA de la FDA pour recevoir l’approbation avant d’aller sur le marché.,
classification des dispositifs médicaux en Europe

Commission européenne
la réglementation d’un dispositif médical dans l’Union Européenne (UE) est établie par les Directives sur les dispositifs médicaux de la Commission européenne (CE).
le chemin vers le marché en Europe est d’obtenir un marquage CE.
pour déterminer ce qui est requis pour obtenir un marquage CE de votre dispositif médical, vous devez d’abord déterminer la classification UE de votre dispositif médical., Le règlement de l’Union européenne sur les dispositifs médicaux (EU MDR) contient les informations nécessaires pour déterminer la classe de votre dispositif.
EU MDR 2017/745 modifie la Directive 2001/83/CE, le règlement (CE) no 178/2002 et le règlement (CE) no 1223/2009 et abroge les Directives 90/385/CEE et 93/42 / CEE du Conseil. EU MDR deviendra le règlement obligatoire pour les dispositifs médicaux à partir de mai 2020.
vous devrez déterminer si votre dispositif médical est:
- Non invasif
- tout dispositif qui ne pénètre pas dans le corps par un orifice ou la surface du corps., Ces appareils sont généralement de classe I; cependant, certaines règles et exceptions s’appliquent qui pourraient les rendre de classe II ou supérieure.
- Envahissantes
- Tout dispositif qui, en tout ou en partie, pénètre à l’intérieur du corps, soit par un orifice du corps ou à travers la surface du corps.
- actif
- tout dispositif dont le fonctionnement dépend d’une source d’énergie autre que celle générée par le corps humain à cet effet, ou par gravité, et qui agit en modifiant la densité de cette énergie ou en la convertissant.,
pour chacune des grandes catégories, certaines règles s’appliquent, décrites à l’annexe VIII du nouveau règlement sur les instruments médicaux. Ces catégories, associées à la durée d’utilisation, rendent la détermination de la classification assez simple.
Par exemple, un appareil utilisé en continu pendant moins de 60 minutes est considéré comme transitoire, 60 minutes à 30 jours est considéré comme à court terme et plus de 30 jours est considéré comme à long terme.,
dans cet esprit, pour déterminer la classification UE de votre appareil, nous pouvons utiliser l’exemple de cathéter percutané utilisé précédemment dans ce guide pour la classification FDA.
disons que je détermine que mon dispositif médical correspond à la catégorie « invasive”; cela réduit ma recherche aux Règles 5, 6, 7 et 8.
je peux ensuite affiner les règles car je sais que mon dispositif médical est à court terme car il est utilisé pour une période supérieure à 24 heures et inférieure à 30 jours.
à partir de là, je suis en mesure de déterminer que la règle 7 est la plus applicable à la classification de mon appareil.,

Source: EU MDR 2017/745 annexe VIII 5.3
étant donné que mon cathéter percutané administrera des médicaments, je peux confirmer que mon dispositif médical est considéré comme un dispositif médical de classe IIb sur le marché européen.
déterminer votre chemin vers le marché en EUROPE
L’Union européenne a un système de classification des produits similaire à celui des États-Unis.,:
- Classe I
- Classe IIa
- Classe IIb
- Classe III
Dans tous les cas pour les dispositifs médicaux à être vendus dans l’Union Européenne, la documentation technique est une étape nécessaire dans le processus d’obtention du Marquage CE. Toutes les classes de dispositifs médicaux dans l’UE nécessitent de travailler avec un organisme notifié, à l’exception de ceux qui sont de classe I et peuvent être auto-certifiés.
les entreprises devront également travailler avec un représentant autorisé pour s’occuper de l’enregistrement des produits en Europe., Les principales Questions de l’article 11 sur les représentants autorisés de la CE peuvent aider à fournir des précisions supplémentaires sur la manière d’obtenir une classification des dispositifs médicaux en Europe.
la Classification des instruments Médicaux au Canada
![]()
Santé Canada
Le règlement sur les instruments médicaux au Canada sont établis par le Gouvernement du Canada et réglementés par Santé Canada.
à l’instar des États-Unis et de l’UE, pour vendre sur le marché Canadien, vous devez d’abord déterminer la classification des instruments médicaux en vertu de la réglementation canadienne.,
semblable aux exigences énoncées dans le RDM de L’UE, Santé Canada fournit une ligne directrice relativement simple et facile à suivre sur le système de Classification fondé sur le risque pour les instruments de Diagnostic non in Vitro que les fabricants d’instruments médicaux peuvent utiliser lorsqu’ils vendent sur ce marché.,
Santé Canada définit quatre groupes d’instruments médicaux diagnostiques non in vitro:
- Instruments invasifs (Règles 1 à 3)
- instruments non invasifs (règles 4 à 7)
- Instruments actifs (règles 8 à 12)
- règles spéciales (règles 13 à 16)
pour chacune des grandes catégories, un ensemble de règles s’applique. Ces règles sont ce que les fabricants devraient suivre afin de déterminer la classification des risques de leur appareil.
je reprendrai l’exemple du cathéter percutané dans le cas de la commercialisation d’un tel appareil au Canada.,
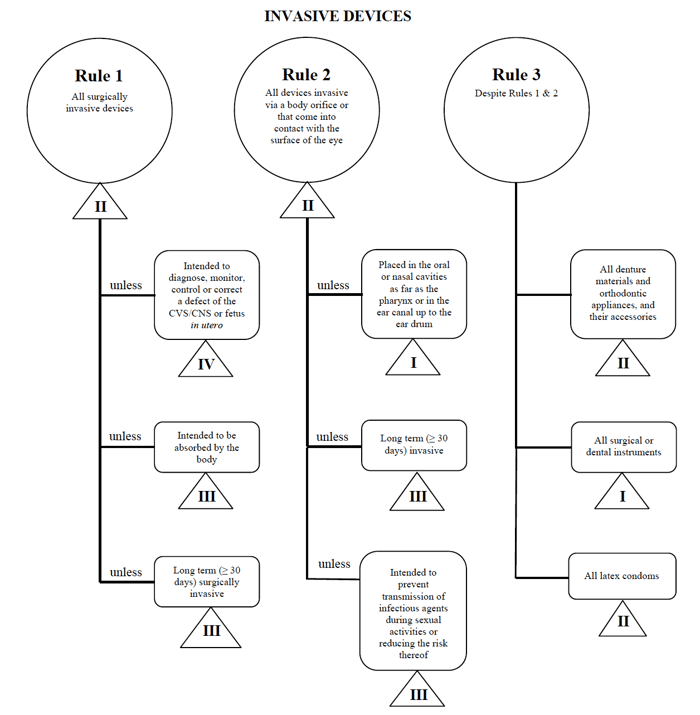
Source: lignes directrices sur le système de Classification fondé sur le risque pour les dispositifs de Diagnostic Non In Vitro
je détermine que mon dispositif médical correspond à la catégorie « invasive”, en réduisant ma recherche aux Règles 1, 2 et 3.
Après avoir examiné les options, je détermine que la Règle 1 s’applique.
selon mon utilisation prévue, mon instrument médical est considéré comme de classe II au Canada.,
déterminer votre voie d’accès au marché au CANADA
Il existe quatre niveaux de classification des instruments médicaux au Canada:
- Classe I
- Classe II
- classe III
- classe IV
avant de mettre sur le marché au Canada, vous devez d’abord Demander une licence d’instrument médical. Les instruments médicaux de classe I ne nécessitent pas de licence. Les fabricants peuvent consulter le document d’orientation de Santé Canada, qui vous guide dans ce processus.,
Les fabricants d’instruments médicaux de classe III et de classe IV peuvent obtenir leur licence en soumettant une demande de mise en marché préalable, dans les formats ToC ou Santé Canada, pour entrer sur le marché canadien.
vous devrez également obtenir la certification ISO 13485 avec MDSAP.
mise à jour du Règlement de Santé Canada: PAIMM
à compter de janvier 2019, tous les fabricants d’instruments médicaux qui vendent des instruments médicaux de classe II et plus sur le marché canadien doivent faire partie du programme D’Audit unique des instruments médicaux (PAIMM).,
ces fabricants doivent subir et réussir un audit complet de leur système de gestion de la qualité (SGQ) dans le cadre du programme au Canada. À l’heure actuelle, 6 régions du monde participent au PAEMD, dont le Canada, les États-Unis, le Japon, Le Brésil et l’Australie.
outre les fabricants requis par Santé Canada pour participer au programme, la participation au PASCMD est facultative pour les fabricants.,
pour obtenir la certification MDSAP, les fabricants d’instruments médicaux doivent suivre les trois étapes suivantes:
- demande et examen
- vérification de la documentation hors site
- vérification sur place
les fabricants d’instruments qui vendent au Canada feront l’objet d’examens annuels, avec un audit de recertification tous les trois ans. La conformité au programme D’Audit unique des dispositifs médicaux repose sur le respect des lignes directrices de la norme ISO 13485 sur les systèmes de management de la qualité des dispositifs médicaux.,
nous vous recommandons de former vos équipes de produits et de qualité et de réglementation aux exigences applicables du MDSAP afin de rationaliser votre application.
notre outil gratuit d’évaluation des lacunes pour MDSAP et ISO 13485 aide les fabricants d’appareils à évaluer les lignes directrices du SMQ de la norme ISO ainsi que les exigences des organismes d’audit de Santé Canada (AO) et d’autres régions participant au programme.,
réflexions finales
Vous avez maintenant les informations et les ressources dont vous avez besoin pour déterminer la classification de votre dispositif médical et le chemin vers le marché dans les trois plus grands marchés du monde.
quelle que soit la classification de votre appareil, il est impératif que vous suiviez les directives réglementaires qui s’appliquent à chaque marché. Respecter la conformité est un aspect clé de la gestion de la qualité qui décidera en fin de compte du sort de votre appareil et de l’entreprise dans son ensemble.,
chez Greenlight Guru, nous apprécions l’importance de la gestion de la qualité des dispositifs médicaux (MDQMS). Notre logiciel MDQMS est la seule solution au monde conçue spécifiquement pour les dispositifs médicaux. Il est aligné sur les dernières pratiques exemplaires standard de l’industrie pour la gestion des contrôles de conception des produits et des risques, ainsi que des activités de contrôle des changements et d’autres événements de qualité, offrant une traçabilité complète tout au long du cycle de vie de votre dispositif médical.
vous recherchez une solution de contrôle de conception pour vous aider à commercialiser plus rapidement des dispositifs médicaux plus sûrs avec moins de risques?, Cliquez ici pour faire un tour rapide du logiciel QMS des dispositifs médicaux de Greenlight Guru →