
to, czym zamierzam się z Tobą podzielić, to przewodnik po klasyfikacji wyrobów medycznych.
w tym przewodniku przedstawiam krok po kroku podejście do określenia, w jaki sposób Twoje urządzenie medyczne zostanie sklasyfikowane przez Amerykańską FDA, Komisję Europejską i Health Canada. Uzyskanie podstawowej wiedzy na temat klasyfikacji produktów zgodnie z przepisami będzie nieocenione dla Twoich wysiłków na rzecz wprowadzania nowych produktów na rynek.,
Klasyfikacja regulacyjna 101
zasady, które mają zastosowanie do wyrobu medycznego, zależą od tego, w jaki sposób produkt jest klasyfikowany przez agencje regulacyjne. Każda agencja regulacyjna zdefiniowała kilka różnych klasyfikacji wyrobów medycznych.
producenci wyrobów medycznych Sprzedający na całym świecie muszą zapoznać się z obowiązującymi przepisami tych rynków. Jest to łatwiej powiedzieć niż zrobić i może być wyzwaniem dla większości producentów., USA mają swój zestaw zasad, podczas gdy Kanada przestrzega innych, a Europa jeszcze innych.
na szczęście istnieje wiele podobieństw między międzynarodowymi przepisami i normami dotyczącymi wyrobów medycznych. Ten przewodnik ma na celu pokazanie, jak klasyfikować urządzenie na różnych rynkach na całym świecie.

dlaczego Klasyfikacja regulacyjna ma znaczenie?
Wiedza o klasyfikacji wyrobu medycznego jest ważna z następujących powodów:
- Klasyfikacja produktu określi, co musisz zrobić, zanim będziesz mógł sprzedać swój produkt.,
- Klasyfikacja produktów pomoże Ci ustalić wymagania na etapie rozwoju produktu, w szczególności kontrole projektu.
- Klasyfikacja produktów jest ważnym elementem w określaniu, ile będzie kosztować wprowadzenie urządzenia na rynek i daje wyobrażenie, jak długo to potrwa.
z tego powodu mam zamiar dostarczyć Ci trochę wskazówek, aby lepiej zrozumieć, co robić i jak to zrobić.,
Uwaga, informacje, które mam zamiar dostarczyć, mają na celu zapoznanie Cię z klasyfikacją regulacyjną wyrobów medycznych i tym, co jest wymagane dla Twojego wyrobu medycznego.
poniższa treść nie jest wyczerpującym przewodnikiem po publikacjach regulacyjnych, ale powinna zawierać podstawowe wskazówki i wskazówki dotyczące identyfikacji, jak ustanowić ścieżkę do rynku.
zostanę przy „wielkiej trójce”, którą powinieneś znać, jeśli chodzi o klasyfikację wyrobów medycznych:
- U. S., Żywności & Drug Administration, Center for Devices & Radiological Health (FDA CDRH)
- Komisja Europejska
- Health Canada
Medical Device Regulatory Classification in the U. S.

U. S. Food & Drug Administration (FDA)
w Stanach Zjednoczonych urządzenia medyczne są regulowane przez żywność & Drug Administration, lub FDA., Specyficzną gałęzią w ramach FDA jest Centrum urządzeń& Radiological Health (CDRH).
misją CDRH jest ochrona i promocja zdrowia publicznego. Innymi słowy, upewnij się, że urządzenia medyczne są bezpieczne. W Stanach Zjednoczonych wyroby medyczne należą do klasy I, II Lub III. klasyfikacja FDA CDRH opiera się przede wszystkim na ryzyku, jakie stwarza wyrób medyczny.
wyroby medyczne klasy i są ogólnie uważane za wyroby niskiego ryzyka, a wyroby medyczne klasy III są postrzegane jako najwyższe ryzyko. Rodzaje wymaganych kontroli zależą od klasyfikacji produktu.,
klasyfikacja jest bezpośrednio związana z przeznaczeniem i wskazaniami do stosowania. Rozróżnienie między tymi terminami jest nieco mylące.
- przeznaczenie jest ogólnym przeznaczeniem wyrobu medycznego lub jego funkcji (co „twierdzisz”, że robi to wyrób medyczny).
- wskazania do stosowania opis choroby lub stanu, który wyrób medyczny będzie diagnozował, leczył, zapobiegał, leczył lub łagodził, w tym opis docelowej populacji pacjentów.
pamiętaj o tym., Przeznaczenie i wskazania do użytkowania wyrobu medycznego wyrażają powód, dla którego wpadł Pan / Pani na pomysł nowego wyrobu medycznego.
Jak znaleźć obowiązujące przepisy FDA dla Twojego wyrobu medycznego
po określeniu przeznaczenia i wskazań do stosowania, teraz musisz znaleźć możliwe przepisy i kody produktów. Śledzenie klasyfikacji regulacyjnej dla Twojego produktu za pośrednictwem FDA zajmuje trochę czasu i wytrwałości.,
nie nudząc cię zbyt wieloma szczegółami, FDA ustanowiła kilka ogólnych kategorii w oparciu o specjalizację medyczną w CFR Tytuł 21 – żywność i leki: części 862 do 892.
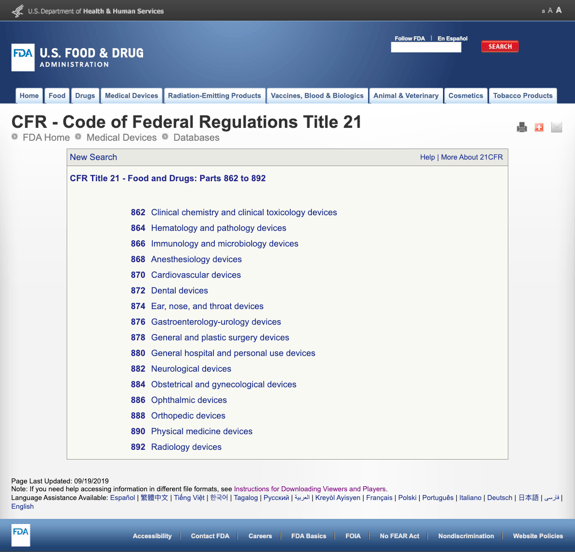
Kiedy znajdziesz możliwe kategorie i klikniesz numer Rozporządzenia FDA, lista możliwości nagle wydaje się nieskończona. Oto częściowy widok opcji dla części 870:

może to być frustrujące i przytłaczające.,
gdy znajdziesz regulację, która wydaje się być możliwa, możesz kliknąć na link i uzyskać więcej szczegółów, aby dokonać ustalenia.
na przykład, jeśli myślę, że moje urządzenie pasuje do cewnika przezskórnego 870.1250, klikam link i otrzymuję tę informację:

Podane szczegóły dają mi pewien pomysł, czy moje przeznaczenie i wskazania do stosowania są zgodne z tą konkretną regulacją. Odkrywam również klasyfikację urządzeń FDA.,
w tym przykładzie dowiaduję się, że mój produkt jest wyrobem medycznym klasy II (standardy wydajności), co oznacza, że będę musiał złożyć 510(k) do FDA przed uzyskaniem odprawy rynkowej. Podzielę się więcej na temat rodzajów zgłoszeń FDA w tym przewodniku.
Next – Znajdź Kody produktów mające zastosowanie do Twojego urządzenia
znalezienie odpowiednich przepisów dotyczących Twojego urządzenia medycznego i klasyfikacji to pierwsza część. Teraz musisz znaleźć odpowiednie kody produktów. Oto jak to zrobić:
przejdź do bazy danych klasyfikacji produktów FDA i wpisz znaleziony numer regulaminu., Jeśli znajdziesz więcej niż jedną możliwość, musisz powtórzyć ten proces dla każdej z nich.
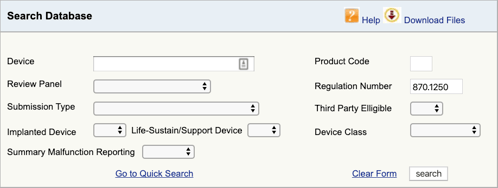
Po kliknięciu „szukaj” otrzymasz listę możliwych kodów produktów.
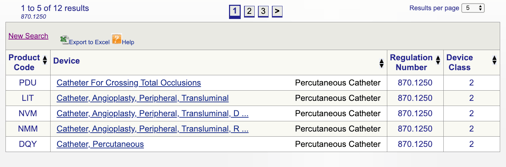
Możesz następnie przejrzeć każdy indywidualny kod, aby określić najlepszą opcję dla Twojego produktu, klikając każdy kod.
określenie Twojej drogi do wprowadzenia na rynek w USA
znajomość obowiązujących przepisów i kodu produktu (jak opisano powyżej) jest niezbędna, aby określić klasyfikację twojego wyrobu medycznego., Po uzyskaniu tych informacji będziesz mógł określić „ścieżkę”, aby zarejestrować produkt w FDA.,
FDA definiuje trzy regulatory kontrolne dla każdej klasy wyrobów medycznych:
- urządzenia medyczne klasy i (o niskim lub umiarkowanym ryzyku): urządzenia medyczne klasy II (o umiarkowanym lub wysokim ryzyku): urządzenia medyczne klasy II (o umiarkowanym lub wysokim ryzyku): urządzenia medyczne klasy III (O WYSOKIM RYZYKU): urządzenia medyczne klasy III (O WYSOKIM RYZYKU): urządzenia medyczne klasy III (O WYSOKIM RYZYKU): urządzenia medyczne klasy III (O WYSOKIM RYZYKU): urządzenia medyczne klasy III (O WYSOKIM RYZYKU)
pozwól, że sprowadzę się do tego:
Jeśli stwierdzisz, że twój produkt jest „wyłączony,” wtedy stosuje się tylko ogólne kontrole i nie jest wymagane formalne złożenie wniosku przez FDA. Musisz jednak zarejestrować swój zakład w FDA, a następnie wymienić produkt.,
Jeśli stwierdzisz, że twój produkt wymaga specjalnych kontroli, oznacza to, że będziesz musiał przygotować zgłoszenie 510 (k) do FDA i otrzymać zezwolenie przed wejściem na rynek. Następnie musisz zarejestrować swój zakład i wymienić produkt.
Jeśli stwierdzisz, że twój produkt wymaga zatwierdzenia przed wprowadzeniem na rynek, oznacza to, że będziesz musiał postępować zgodnie z procesem FDA PMA, aby otrzymać zatwierdzenie przed wejściem na rynek.,
Klasyfikacja Wyrobów Medycznych w Europie

Komisja Europejska
przepisy dotyczące wyrobów medycznych w Unii Europejskiej (UE) są ustanawiane na podstawie dyrektyw Komisji Europejskiej (we).
drogą do wprowadzenia na rynek w Europie jest uzyskanie oznakowania CE.
aby dowiedzieć się, co jest wymagane do uzyskania oznakowania CE wyrobu medycznego, należy najpierw określić unijną klasyfikację wyrobu medycznego., Rozporządzenie Unii Europejskiej dotyczące wyrobów medycznych (EU MDR) zawiera informacje niezbędne do określenia klasy urządzenia.
EU MDR 2017/745 zmienia dyrektywę 2001/83/WE, Rozporządzenie (WE) nr 178/2002 i rozporządzenie (WE) nr 1223/2009 oraz uchylające dyrektywy Rady 90/385/EWG i 93/42 / EWG. Dyrektywa MDR UE stanie się obowiązkowym rozporządzeniem dla wyrobów medycznych począwszy od maja 2020 r.
musisz określić, czy Twoje urządzenie medyczne to:
- nieinwazyjne
- jakiekolwiek urządzenie, które nie przenika przez otwór lub powierzchnię ciała., Urządzenia te zazwyczaj należą do klasy I, jednak obowiązują pewne zasady i wyjątki, które mogą uczynić je klasą II lub wyższą.
- inwazyjne
- dowolne urządzenie, które w całości lub w części wnika do wnętrza ciała, albo przez otwór ciała, albo przez powierzchnię ciała.
- aktywne
- każde urządzenie, którego działanie zależy od źródła energii innego niż generowane w tym celu przez ludzkie ciało lub grawitację, i które działa poprzez zmianę gęstości lub konwersję tej energii.,
dla każdej z szerokich kategorii obowiązują pewne zasady określone w załączniku VIII do nowego rozporządzenia w sprawie wyrobów medycznych. Kategorie te w połączeniu z czasem ich stosowania sprawiają, że ustalenie klasyfikacji jest dość proste.
na przykład urządzenie w ciągłym użytkowaniu przez mniej niż 60 minut jest uważane za krótkotrwałe, 60 minut do 30 dni jest uważane za krótkotrwałe, a ponad 30 dni za długoterminowe.,
Mając to na uwadze, aby określić klasyfikację Twojego urządzenia w UE, możemy użyć przykładu cewnika przezskórnego stosowanego wcześniej w tym przewodniku do klasyfikacji FDA.
powiedzmy, że ustalam, że moje urządzenie medyczne pasuje do kategorii „inwazyjnej”; to zawęża moje wyszukiwanie do zasad 5, 6, 7 i 8.
mogę zawęzić zasady, ponieważ Wiem, że moje urządzenie medyczne jest krótkotrwałe, ponieważ jest używane przez okres dłuższy niż 24 godziny i mniej niż 30 dni.
stamtąd jestem w stanie stwierdzić, że zasada 7 jest najbardziej odpowiednia do klasyfikacji mojego urządzenia.,

źródło: EU MDR 2017/745 aneks VIII 5.3
ponieważ mój cewnik przezskórny będzie podawać leki, mogę potwierdzić, że moje wyrób medyczny jest uważany za wyrób medyczny klasy IIb na rynku europejskim.
,:
- Klasa I
- Klasa IIa
- Klasa IIb
- Klasa III
we wszystkich przypadkach wyrobów medycznych przeznaczonych do sprzedaży na terenie Unii Europejskiej wymagana jest dokumentacja techniczna w procesie uzyskiwania oznakowania CE. Wszystkie klasy wyrobów medycznych w UE wymagają współpracy z jednostką notyfikowaną, z wyjątkiem tych, które należą do klasy I i mogą mieć własną certyfikację.
firmy będą również musiały współpracować z Autoryzowanym Przedstawicielem, aby zająć się rejestracją produktów w Europie., Najważniejsze pytania zawarte w art. 11 dotyczące upoważnionych przedstawicieli we mogą pomóc w doprecyzowaniu sposobów osiągnięcia klasyfikacji wyrobów medycznych w Europie.
Klasyfikacja Wyrobów Medycznych w Kanadzie
![]()
Health Canada
przepisy dotyczące wyrobów medycznych w Kanadzie są ustanawiane przez rząd Kanady i regulowane przez Health Canada.
podobnie jak w USA i UE, aby sprzedawać na rynku kanadyjskim, musisz najpierw określić klasyfikację wyrobów medycznych zgodnie z kanadyjskim rozporządzeniem.,
podobnie jak wymogi określone w MDR UE, Health Canada zapewnia dość proste i łatwe do naśladowania wytyczne dotyczące systemu klasyfikacji opartej na ryzyku wyrobów do diagnostyki in Vitro dla producentów wyrobów medycznych do stosowania przy sprzedaży na tym rynku.,
Health Canada definiuje cztery grupy nieinwazyjnych wyrobów medycznych:
- wyrobów inwazyjnych (zasady 1 – 3)
- wyrobów nieinwazyjnych (Zasady 4-7)
- wyrobów aktywnych (Zasady 8 – 12)
- specjalnych reguł (Zasady 13 – 16)
dla każdej z szerokich kategorii obowiązuje zestaw reguł. Zasady te są tym, czego powinni przestrzegać producenci, aby określić klasyfikację ryzyka swojego wyrobu.
jeszcze raz użyję przykładu cewnika przezskórnego w przypadku sprzedaży takiego urządzenia w Kanadzie.,
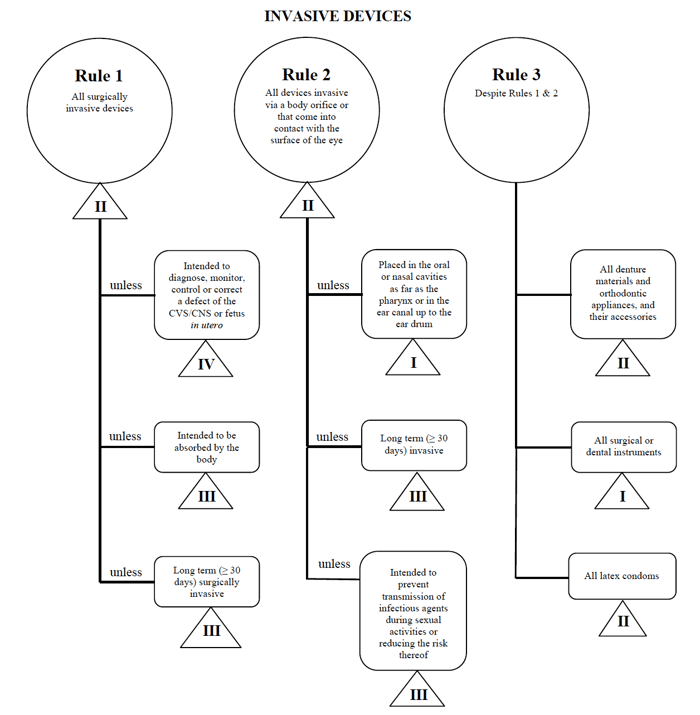
źródło: Guidance on the Risk based Classification System for Non-in Vitro Diagnostic Devices
stwierdzam, że mój wyrób medyczny pasuje do kategorii „inwazyjnej”, zawężając moje wyszukiwanie do reguł 1, 2 i 3.
po przejrzeniu opcji stwierdzam, że zasada 1 ma zastosowanie.
w oparciu o moje przeznaczenie, moje urządzenie medyczne jest uważane za II klasę w Kanadzie.,
określanie Twojej drogi do wprowadzenia na rynek w Kanadzie
w Kanadzie istnieją cztery poziomy klasyfikacji wyrobów medycznych:
- Klasa I
- klasa II
- klasa III
- klasa IV
przed wejściem na rynek w Kanadzie musisz najpierw złożyć wniosek o licencję na wyroby medyczne. Wyroby medyczne klasy I nie wymagają licencji. Producenci mogą odwołać się do wytycznych Health Canada, które przeprowadzą cię przez ten proces.,
producenci wyrobów medycznych klasy III I IV mogą otrzymać licencję, składając przedmarketowy wniosek, w formacie ToC lub Health Canada, o wejście na rynek kanadyjski.
musisz również uzyskać certyfikat ISO 13485 z MDSAP.
aktualizacja przepisów Health Canada: Mdsap
od stycznia 2019 r.wszyscy producenci wyrobów medycznych Sprzedający wyroby medyczne klasy II i wyższej na rynek kanadyjski muszą być częścią programu pojedynczego audytu Wyrobów Medycznych (Mdsap).,
producenci ci muszą przejść i przejść pełny audyt swojego systemu zarządzania jakością (QMS) w ramach programu w Kanadzie. Obecnie w MDSAP uczestniczy 6 regionów na całym świecie, w tym Kanada, USA, Japonia, Brazylia i Australia.
poza producentami wymaganymi przez Health Canada do udziału w programie, udział w MDSAP jest opcjonalny dla producentów.,
aby uzyskać certyfikat MDSAP, producenci urządzeń medycznych muszą wykonać następujące trzy kroki:
- Application and review
- audyt dokumentacji zewnętrznej
- audyt na miejscu
producenci urządzeń sprzedających do Kanady będą poddawani corocznym przeglądom, z audytem recertyfikacyjnym co trzy lata. Zgodność z programem pojedynczego audytu Wyrobów Medycznych opiera się na spełnieniu wytycznych normy ISO 13485 dotyczącej systemów zarządzania jakością wyrobów medycznych.,
zalecamy przeszkolenie zespołów ds. produktów, jakości i przepisów w zakresie obowiązujących wymagań MDSAP w celu usprawnienia aplikacji.
nasze bezpłatne narzędzie do oceny luk dla MDSAP i ISO 13485 pomaga producentom urządzeń ocenić wytyczne QMS z normy ISO wraz z wymaganiami organizacji Audytujących Health Canada (ao) i innych regionów uczestniczących w programie.,
myśli końcowe
masz teraz informacje i zasoby potrzebne do określenia klasyfikacji wyrobów medycznych i ścieżki do wprowadzenia na rynek w trzech największych marketach na całym świecie.
niezależnie od klasyfikacji urządzenia, konieczne jest przestrzeganie wytycznych regulacyjnych obowiązujących na każdym rynku. Zgodność z przepisami jest kluczowym aspektem zarządzania jakością, który ostatecznie zadecyduje o losie urządzenia i firmy jako całości.,
w Greenlight Guru cenimy sobie znaczenie zarządzania jakością urządzeń medycznych (MDQMS). Nasze oprogramowanie MDQMS jest jedynym na świecie rozwiązaniem zaprojektowanym specjalnie dla urządzeń medycznych. Jest zgodny z najnowszymi standardami branżowymi, najlepszymi praktykami w zakresie zarządzania kontrolą projektu produktu i ryzykiem, a także działaniami kontrolnymi zmian i innymi zdarzeniami o wysokiej jakości, zapewniając pełną identyfikowalność w całym cyklu życia urządzenia medycznego.
szukasz rozwiązania do kontroli projektu, które pomoże Ci szybciej wprowadzić bezpieczniejsze urządzenia medyczne na rynek przy mniejszym ryzyku?, Kliknij tutaj, aby szybko zapoznać się z oprogramowaniem QMS urządzenia medycznego Greenlight Guru →