
vad jag ska dela med dig är en guide till medicinsk enhet reglerande klassificering.
i den här guiden kommer jag att ge dig en steg-för-steg-metod för att bestämma hur din medicinska enhet kommer att klassificeras av amerikanska FDA, Europeiska kommissionen och Health Canada. Att få en grundläggande förståelse för reglerande produktklassificering kommer att vara ovärderlig för dina ansträngningar att få nya produkter på marknaden.,
regulatory Classification 101
de regler som gäller för din medicinska enhet beror på hur din produkt klassificeras av tillsynsmyndigheterna. Varje tillsynsmyndighet har definierat flera olika klassificeringar för medicintekniska produkter.
klassificeringarna är för det mesta eller som en allmän regel relaterade till den upplevda risken för produkttypen.
tillverkare av medicintekniska produkter som säljer internationellt måste bekanta sig med gällande regler på dessa marknader. Detta är lättare sagt än gjort och kan vara en utmaning för de flesta tillverkare., USA har sin uppsättning regler, medan Kanada följer en annan, och Europa en annan fortfarande.
lyckligtvis finns det många paralleller mellan internationella medicintekniska föreskrifter och standarder. Den här guiden är utformad för att visa dig hur du klassificerar din enhet på olika marknader runt om i världen.

Varför spelar reglerande klassificering ens någon roll?
veta hur din medicinska enhet klassificeras frågor av följande skäl:
- produktklassificering kommer att avgöra vad du måste göra innan du kan sälja din produkt.,
- produktklassificering hjälper dig att fastställa krav under produktutvecklingsfasen, särskilt konstruktionskontroller.
- produktklassificering är en viktig komponent för att bestämma hur mycket det kommer att kosta att få din enhet att marknadsföra och ge dig en uppfattning om hur lång tid det tar.
På grund av detta kommer jag att ge dig lite vägledning för att bättre förstå vad du ska göra och hur du gör det.,
Observera att den information jag ska tillhandahålla är avsedd att hjälpa dig att utbilda dig om medicinsk apparatklassificering och vad som krävs för din medicinska enhet.
följande innehåll är inte en omfattande guide till regulatoriska inlagor, men bör ge dig några grundläggande vägledning och riktning för att identifiera hur man etablerar väg till marknaden.
Jag håller mig till ”big 3” Du borde veta när det gäller klassificering av medicinsk enhet:
- US, Food & Drug Administration, Center for Devices & radiologisk hälsa (FDA CDRH)
- Europeiska kommissionen
- hälsa Kanada
medicinsk enhet regulatorisk klassificering i USA

amerikansk mat & div id=”754dc4608a”>Drug Administration (FDA)
i USA regleras medicintekniska produkter av maten& Drug Administration, eller FDA., Den specifika grenen inom FDA är Centrum för enheter & radiologisk hälsa (CDRH).CDRH: s uppdrag är att skydda och främja folkhälsan . Med andra ord, se till att medicintekniska produkter är säkra. I USA är medicintekniska produkter antingen klass i, klass II eller klass III. FDA CDRH-klassificeringen baseras främst på risk som den medicinska enheten utgör.
medicintekniska produkter av klass I anses generellt vara lågriskprodukter och medicintekniska produkter av klass III ses som den högsta risken. Vilka typer av kontroller som krävs beror på din produkts klassificering.,
klassificeringen är direkt relaterad till Avsedd användning och indikationer för användning. Skillnaden mellan dessa termer är lite förvirrande.
- Avsedd användning är det allmänna syftet med den medicinska enheten eller dess funktion (vad du ”hävdar” den medicinska enheten gör).
- indikationer för användning beskriv sjukdomen eller tillståndet den medicinska produkten kommer att diagnostisera, behandla, förebygga, bota eller mildra, inklusive en beskrivning av målpatientpopulationen.
Kom ihåg detta., Den avsedda användningen och indikationerna för användning av din medicinska enhet uttrycker anledningen till att du hade denna idé för en ny medicinsk enhet.
så här hittar du tillämpliga FDA-föreskrifter för din medicinska enhet
När du har definierat avsedd användning och indikationer för användning måste du nu hitta möjliga regler och produktkoder. Att spåra regleringsklassificering för din produkt via FDA tar lite tid och uthållighet.,
utan att tråkiga dig med för många detaljer har FDA etablerat flera allmänna kategorier baserat på den medicinska specialiteten i CFR Titel 21 – mat och droger: delar 862 till 892.
När du hittar de möjliga kategorierna och klickar på FDA-regelnumret verkar listan över möjligheter plötsligt oändlig. Här är en partiell bild av alternativen för del 870 kardiovaskulära enheter:

detta kan vara frustrerande och överväldigande.,
När du hittar en förordning som verkar vara en möjlig passform, kan du klicka på länken och få mer information för att göra en beslutsamhet.
till exempel, om jag tror att min enhet passar i ”870.1250 perkutan kateter”, klickar jag på länken och får den här informationen:

de uppgifter som tillhandahålls ger mig en aning om min avsedda användning och indikationer för användning stämmer överens med denna specifika förordning. Jag upptäcker också FDA-enhetens klassificering.,
i det här exemplet lär jag mig att min produkt är en klass II-medicinsk enhet (prestandastandarder), vilket innebär att jag måste skicka in en 510(k) till FDA innan jag får marknadsgodkännande. Jag delar mer om typer av FDA inlagor vidare i denna guide.
Nästa-hitta de Produktkoder som gäller för din enhet
att hitta den tillämpliga förordningen för dig medicinsk enhet och klassificering är den första delen. Nu måste du hitta de tillämpliga produktkoderna. Så här:
gå till FDA: s Produktklassificeringsdatabas och skriv in det regleringsnummer du hittade., Om du hittar mer än en möjlighet, måste du upprepa denna process för varje.
När du klickar på ”SÖK” får du en lista över möjliga produktkoder.
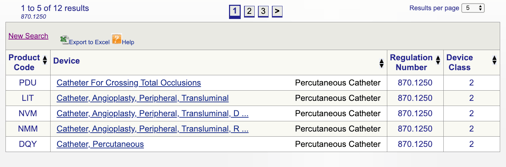
Du kan sedan granska varje enskild kod för att bestämma det bästa alternativet för din produkt genom att klicka på varje kod.
det är nödvändigt att bestämma din väg till marknaden i USA
att känna till tillämplig reglering och Produktkod (enligt beskrivningen ovan) för att du ska kunna bestämma klassificeringen av din medicinska enhet., När du har denna information kommer du nu att kunna bestämma ”sökvägen” för att få din produkt registrerad hos FDA.,
FDA definierar tre regulatoriska kontroller för varje medicinsk enhet klass:
- klass i medicinsk enhet (låg till måttlig risk): allmänna kontroller
- klass II medicinsk enhet (måttlig till hög risk): allmänna kontroller och särskilda kontroller
- klass III medicinsk enhet (hög risk): allmänna kontroller och förhandsgodkännande (PMA)
Låt mig koka ner det till detta:
om du hittar att din produkt är ”undantagen” då gäller endast allmänna kontroller och ingen formell FDA-inlämning krävs. Du behöver dock registrera din anläggning hos FDA och sedan lista produkten.,
om du hittar din produkt kräver särskilda kontroller, detta innebär att du måste förbereda en 510(k) inlämning till FDA och få clearance innan du går till marknaden. Därefter måste du registrera din anläggning och lista produkten.
om du hittar din produkt kräver premarket godkännande, detta innebär att du måste följa FDA PMA processen för att få godkännande innan du går till marknaden.,
klassificering av medicintekniska produkter i Europa

Europeiska kommissionen
reglerna för medicintekniska produkter i Europeiska unionen (EU) fastställs genom direktiven om medicintekniska produkter av Europeiska kommissionen (EG).
vägen till marknaden i Europa är att få en CE-märkning.
för att ta reda på vad som krävs för att få en CE-märkning av din medicintekniska produkt måste du först bestämma EU: s klassificering av din medicintekniska produkt., EU: s förordning om medicintekniska produkter (EU MDR) innehåller den information som behövs för att bestämma din enhetsklass.
EU MDR 2017/745 ändrar direktiv 2001/83/EG, förordning (EG) nr 178/2002 och förordning (EG) nr 1223/2009 och upphäver rådets direktiv 90/385/EEG och 93/42 / EEG. EU: s MDR kommer att bli den obligatoriska förordningen för medicintekniska produkter från och med maj 2020.
Du måste avgöra om din medicinska enhet är:
- icke-invasiv
- alla enheter som inte tränger in i kroppen genom en öppning eller kroppsytan., Dessa enheter är vanligtvis klass I; Dock gäller vissa regler och undantag som kan göra dem klass II eller högre.
- invasiv
- alla enheter som helt eller delvis tränger in i kroppen, antingen genom en kroppsöppning eller genom kroppens yta.
- aktiv
- alla enheter vars funktion beror på en annan energikälla än den som genereras av människokroppen för detta ändamål, eller genom gravitation, och som verkar genom att ändra densiteten hos eller omvandla den energin.,
för var och en av de breda kategorierna finns det vissa regler som gäller, som beskrivs i bilaga VIII till den nya förordningen om medicintekniska produkter. Dessa kategorier tillsammans med användningstiden gör det ganska enkelt att bestämma klassificeringen.
till exempel anses en enhet i kontinuerlig användning i under 60 minuter vara övergående varaktighet, 60 minuter till 30 dagar anses vara kortvarig och över 30 dagar anses vara långsiktig.,
med detta i åtanke, för att bestämma EU: s klassificering av din enhet, kan vi använda det perkutan kateter exempel som används tidigare i denna guide för FDA klassificering.
låt oss säga att jag bestämmer att min medicinska enhet passar in i kategorin” invasiv”; detta begränsar min sökning till reglerna 5, 6, 7 och 8.
Jag kan sedan begränsa reglerna längre eftersom jag vet att min medicintekniska produkt är kortsiktig eftersom den används under en period som är större än 24 timmar och mindre än 30 dagar.
därifrån kan jag bestämma att regel 7 är mest tillämplig på klassificeringen av min enhet.,

källa: EU MDR 2017/745 bilaga VIII 5.3
eftersom min perkutan kateter administrerar läkemedel kan jag bekräfta att min medicintekniska produkt anses vara en klass IIb-medicinteknisk produkt på den europeiska marknaden.
bestämma din väg till marknaden i Europa
Europeiska unionen har ett liknande produktklassificeringssystem som USA,:
- klass i
- klass IIa
- klass IIb
- klass III
i samtliga fall för medicintekniska produkter som ska säljas i Europeiska unionen är teknisk dokumentation ett nödvändigt steg i processen för att erhålla CE-märkning. Alla klasser av medicintekniska produkter i EU kräver samarbete med ett anmält organ, förutom de som är klass I och kan vara självcertifierade.
företag kommer också att behöva arbeta med en auktoriserad representant för att ta hand om produktregistrering i Europa., Artikel 11 Toppfrågor om EG auktoriserade representanter kan bidra till att ge ytterligare klargöranden om hur man uppnår klassificering av medicintekniska produkter i Europa.
klassificering av medicintekniska produkter i Kanada
![]()
Health Canada
reglerna för medicintekniska produkter i Kanada är fastställda av Kanadas regering och reglerade av Health Canada.
I likhet med USA och EU måste du först bestämma klassificeringen av medicintekniska produkter Enligt Kanadas förordning för att sälja till den kanadensiska marknaden.,
i likhet med kraven i EU MDR ger Health Canada en ganska enkel och lätt att följa vägledning om det riskbaserade klassificeringssystemet för icke-in Vitro-diagnostiska produkter för tillverkare av medicintekniska produkter som ska användas vid försäljning till denna marknad.,
Health Canada definierar fyra grupper av icke-in vitro-diagnostiska medicinska enheter:
- invasiva enheter (regler 1-3)
- icke – invasiva enheter (regler 4-7)
- Aktiva enheter (regler 8-12)
- särskilda regler (regler 13-16)
För var och en av de breda kategorierna finns det en uppsättning regler som gäller. Dessa regler är vad tillverkarna bör följa för att bestämma riskklassificeringen av deras enhet.
Jag använder exemplet perkutan kateter En gång till vid marknadsföring av en sådan enhet i Kanada.,
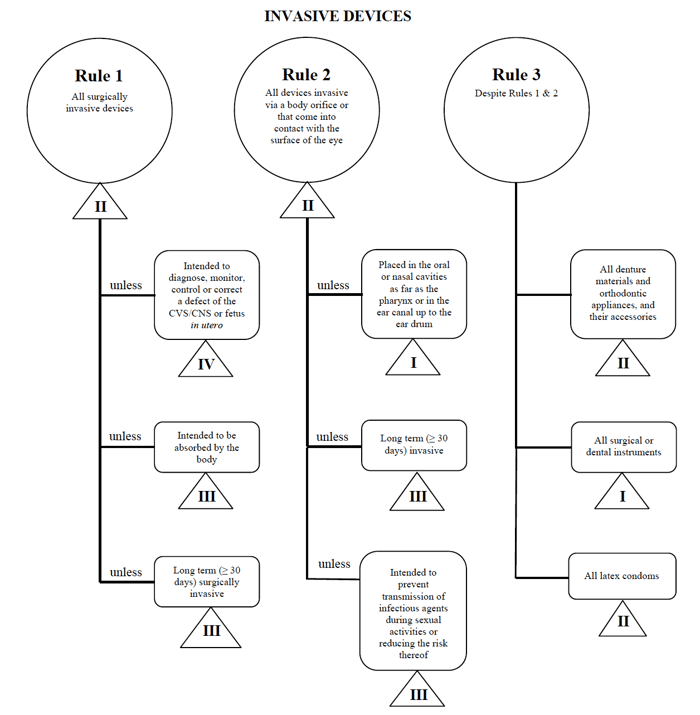
Source: Guidance on the Risk based Classification System for Non-in Vitro Diagnostic Devices
jag bestämmer att min medicinska enhet passar in i kategorin ”invasive”, vilket begränsar min sökning till reglerna 1, 2 och 3.
Efter att ha granskat alternativen bestämmer jag att Regel 1 gäller.
baserat på min avsedda användning anses min medicinska enhet klass II i Kanada.,
bestämma din väg till marknaden i Kanada
det finns fyra nivåer av medicinska enhet klassificeringar i Kanada:
- klass i
- klass II
- klass III
- klass IV
innan du går till marknaden i Kanada, måste du först ansöka om en medicinsk enhet licens. Klass i medicintekniska produkter kräver ingen licens. Tillverkare kan referera till Health Canada guidance document, som går igenom denna process.,
Tillverkare av Klass III och Klass IV medicinsk utrustning kan få sin licens genom att skicka in en premarket ansökan, antingen i Innehållsförteckningen eller Hälsa Kanada format, för att ange den Kanadensiska marknaden.
Du måste också få ISO 13485-certifiering med MDSAP.
Uppdatera för att Hälsa Kanada Förordningar: MDSAP
från och med januari 2019, alla medicintekniska tillverkare som säljer medicintekniska produkter i Klass II och högre till den Kanadensiska marknaden, måste vara en del av den Medicinska Enheten Enda Audit Programme (MDSAP).,
dessa tillverkare måste genomgå och genomgå en fullständig granskning av sitt kvalitetsstyrningssystem (QMS) genom programmet i Kanada. Det finns för närvarande 6 regioner runt om i världen som deltar i MDSAP, inklusive Kanada, USA, Japan, Brasilien och Australien.
förutom de tillverkare som krävs av Health Canada för att delta i programmet är deltagande i MDSAP frivilligt för tillverkare.,
för att få mdsap-certifiering måste tillverkare av medicintekniska produkter slutföra följande tre steg:
- ansökan och översynen
- granskning av dokumentation utanför webbplatsen
- revision på plats
enhetstillverkare som säljer till Kanada kommer att bli föremål för årliga recensioner, med en omcertifieringsrevision var tredje år. Överensstämmelse med Enhetsrevisionsprogrammet för medicintekniska produkter bygger på att följa riktlinjerna i ISO 13485-standarden om kvalitetsledningssystem för medicintekniska produkter.,
Vi rekommenderar att du utbildar dina produkt-och kvalitets-och regleringsteam i tillämpliga MDSAP-krav för att effektivisera din ansökan.
vårt gratis gap assessment tool för mdsap och ISO 13485 hjälper enhetstillverkare att bedöma QMS-riktlinjerna från ISO-standarden tillsammans med kraven från Health Canada auditing organizations (AO) och andra regioner som deltar i programmet.,
slutliga tankar
Du har nu den information och resurser du behöver för att bestämma din klassificering av medicintekniska produkter och väg till marknaden på de tre största marknadsplatserna runt om i världen.
oavsett enhetens klassificering är det absolut nödvändigt att du följer de regler som gäller på varje marknad. Meeting compliance är en viktig aspekt av kvalitetsstyrning som i slutändan bestämmer ödet för din enhet och företag som helhet.,
på Greenlight Guru värdesätter vi vikten av medicinsk kvalitetshantering (MDQMS). Våra MDQMS programvara är den enda lösningen i världen som utformats speciellt för medicintekniska produkter. Den är anpassad till den senaste branschstandarden bästa praxis för hantering av produktdesignkontroller och risker, samt förändringskontrollaktiviteter och andra kvalitetshändelser, vilket ger full spårbarhet under hela livscykeln för din medicinska enhet.
letar du efter en designkontrolllösning som hjälper dig att få säkrare medicintekniska produkter att marknadsföra snabbare med mindre risk?, Klicka här för att ta en snabb rundtur i Greenlight Guru medicinska enhet QMS software →