
내가 무엇을 공유하는 것을 대략한 안내입니다 의학 장치 규제 분류 수 있습니다.
이 안내서에서는 내가를 제공할 것입니다 단계별 접근 방법을 결정하기 위한 귀하의 의료 장치를 분류하여 미국 FDA,유럽위원회,그리고 캐나다 보건부. 을 얻기에 대한 기본적인 이해 규제 제품 분류 헤아릴 수 없을 것이다 당신의 노력을 가지고 새로운 제품을 시장이다.,
규제 분류 101
는 규칙을 적용하여 귀하의 의료 장치에 따라 어떻게 귀하의 제품에 의해 규제 기관입니다. 각 규제 기관은 의료 기기에 대한 몇 가지 다른 분류를 정의했습니다.
분류는 대부분 또는 일반적으로 제품 유형의 인식 된 위험과 관련이 있습니다.
국제적으로 판매하는 의료 기기 제조업체는 해당 시장의 해당 규정을 숙지해야합니다. 이것은 완료보다 쉽게 말하며 대부분의 제조업체에게 도전이 될 수 있습니다., 미국은 일련의 규칙을 가지고 있지만 캐나다는 다른 규칙을 고수하고 유럽은 여전히 다른 규칙을 고수합니다.
다행스럽게도 국제 의료 기기 규정과 표준 사이에는 많은 유사점이 있습니다. 이 가이드는 전 세계의 다른 시장에서 장치를 분류하는 방법을 보여주기 위해 고안되었습니다.

규제 분류가 중요한 이유는 무엇입니까?
는 방법을 알고 귀하의 의료 장치를 분류한 문제는 다음과 같은 장점이 있습니다.
- 제품 분류는 것이 무엇인지 결정하기 전에 할 수 있는 판매할 수 있는 제품입니다.,
- 제품 분류는 제품 개발 단계에서 요구 사항을 수립하고 특히 컨트롤을 설계하는 데 도움이됩니다.
- 제품 분류에 있는 중요한 요소를 결정에 얼마나 많은 비용을 가지 장치를 시장과 몇 가지 아이디어를 줄 그것이 얼마나 오래 걸릴 것입니다.
이 때문에 수행 할 작업과 수행 방법을 더 잘 이해할 수 있도록 약간의 지침을 제공하겠습니다.,
주한 정보가 제공하는 것을 교육하에서 의학 장치 규제 분류 및기 위해 필요한 것은 귀하의 의료 장치입니다.
는 다음과 같은 콘텐츠는 포괄적인 규제 가이드 제출,아직 당신을 제공해야 몇 가지 기본적인 지도와 방향을 식별하는 방법을 설정한 경로를 시장이다.
나는 의료 기기 분류에 관해서 당신이 알아야 할”빅 3″에 충실 할 것이다:
- 미국, 식품&Drug Administration,Center for 장치&방사선의 건강(FDA CDRH)
- 유럽위원회
- 캐나다 보건부
의학 장치 규제 분류에서 미국

미국 식품&DRUG ADMINISTRATION(FDA)
미국에서 의학 장치 규제에 의해 음식을&의약품 관리,또는 FDA., FDA 내의 특정 지점은 Center For Devices&방사선 건강(CDRH)입니다.
CDRH 의 임무는 공중 보건을 보호하고 증진시키는 것입니다. 즉,의료 기기가 안전한지 확인하십시오. 미국에서는 의료 기기가 클래스 I,클래스 II 또는 클래스 III 중 하나입니다.FDA CDRH 분류는 주로 의료 기기가 제기하는 위험에 기반합니다.
클래스 I 의학 장치는 일반적으로 낮은 것으로 간주되는 경우 신원 및 위험 클래스 III 의학 장치로 볼 수 있습니다 가장 높은 위험이 있습니다. 필요한 컨트롤 유형은 제품의 분류에 따라 다릅니다.,
분류는 의도 된 사용 및 사용에 대한 표시와 직접 관련이 있습니다. 이 용어들 사이의 구별은 약간 혼란 스럽습니다.
- 의도 된 사용은 의료 기기 또는 그 기능(귀하가 의료 기기가하는”주장”)의 일반적인 목적입니다.
- 사용 설명하는 질병 또는 상태의 의료 장치를 진단,치료,예방,치료,또는 완화시키기 위해 설명을 포함한의 대상 환자니다.
이것을 명심하십시오., 사용 용도와 표시의 사용에 대한 귀하의 의료 장치를 표현하는 이유는 당신이에 대한 아이디어로 새로운 의학 장치입니다.
는 방법을 찾기 위해 해당 FDA 규정에 대한 귀하의 의료 기기
를 정의하면 사용 용도와 사용을 위한 표시,지금 당신은 당신을 찾아야 해요 가능한 규정 및 제품 코드들이 모체 제품 코드. FDA 를 통해 제품에 대한 규제 분류를 추적하는 데는 약간의 시간과 인내가 필요합니다.,
지루하지 않고 당신은 너무 많은 정보 FDA 설립했다 여러 가지 일반적인 종류에 기반한 의료 전문 Title21CFR-식약:부분 862 892.
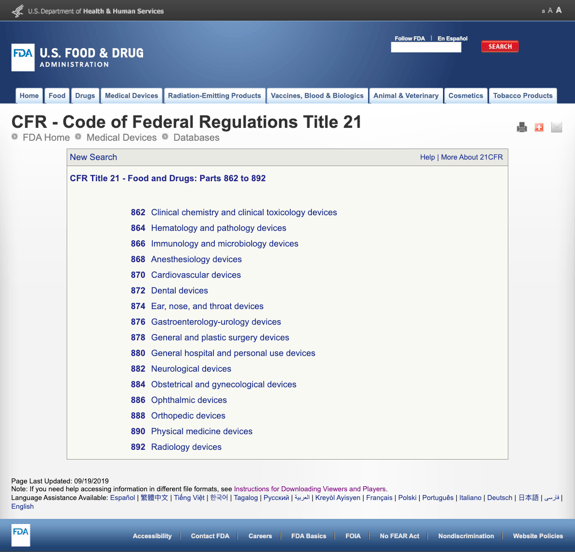
을 찾을 때 가능한 카테고리 클릭 FDA 규정 번호 목록의 가능성은 갑자기 보이 끝이 없습니다. 여기에 부분적으로 옵션에 대한 부분 870 심장 혈관 장치:

이것은 복잡할 수 있습 압도적이다.,
가능한 적합으로 보이는 규정을 찾으면 링크를 클릭하고 자세한 내용을 입수하여 결정을 내릴 수 있습니다.
경우,예를 들어 내는 장치에 맞는”870.1250 매몰 카테테르는”나는 링크를 클릭하고 이 정보를 얻을:

정보 제공하는 나에게 어떤 생각하는 경우 내가 사용 용도와 사용에 대한 징후와 맞이 특정 규정. 나는 또한 FDA 장치 분류를 발견한다.,
이 예에서 나는 배울 수 있는 제품입니다 Class II 의료기기(성능 표준)의미하는 것이 필요하여 제출 510(k)FDA 을 받기 전에 시장니다. 나는이 가이드에 더 FDA 제출의 유형에 대한 자세한 내용을 공유.
Next–찾 제품 코드들이 모체 제품 코드에 해당하는 기기
을 찾는 적용 가능한 규정은 당신을 위해 의료 장치 및 분류 첫 번째 부분입니다. 이제 해당 제품 코드를 찾아야합니다. 방법은 다음과 같습니다.
FDA 제품 분류 데이터베이스로 이동하여 찾은 규정 번호를 입력하십시오., 하나 이상의 가능성을 발견하면 각각에 대해이 과정을 반복해야합니다.
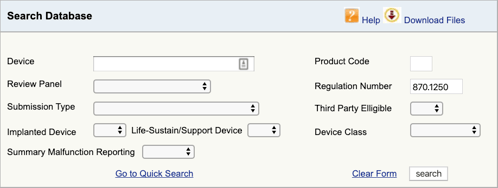
을 클릭하면”검색”,당신의 목록을 얻을 것이 가능한 제품 코드들이 모체 제품 코드.
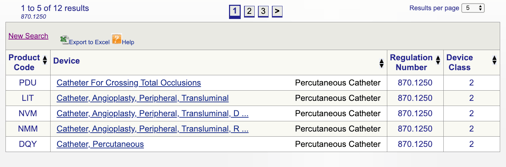
을 검토한 다음 각각의 개별 코드를 결정에 가장 적합한 옵션을 클릭하여 제품에서 각각의 코드입니다.
을 결정하는 경로를 시장에서 미국
아는 적용 가능한 규정 및 제품 코드(위에서 설명한대로)이 필요 당신이 결정하의 분류는 당신의 의료 장치입니다., 일단 당신이 이 정보가 있으면,당신은 지금 FDA 에 등록된 당신의 제품을 얻는”경로”를 결정할 수 있을 것입니다.,
FDA 을 정의 세 가지 규제에 대한 각각의 의료 장치의 종류:
- 클래스 I 의료장치(중간에 낮은 위험):일반 컨트롤
- Class II 의료 장치(적당한 위험이 높은):일반적인 제어 및 특수한 컨트롤
- 클래스 III 의료 장치(고위험):일반적인 제어 및 시판 승인(PMA)
자 나에게 물을 끓이:
찾은 경우에는 제품입니다”면제하고,”다만 일반적인 컨트롤이 적용되며 공식적인 FDA 제출이 필요합니다. 당신은,그러나,FDA 에 당신의 설립을 등록하고 그 후에 제품을 목록으로 만들 필요가 있습니다.,
경우에 당신은 당신의 제품을 찾을 필요로 특수 제어,이미 준비해야 할 것입니다 510(k)제출 FDA 및 허가를받을 하기 전에 시장이다. 그 후에,당신은 당신의 설립을 등록하고 제품을 나열해야합니다.
경우에 당신은 당신의 제품을 찾을 필요합 시판 승인,이것은 당신을 사용하지 않습니다 FDA PMA 프로세스를 승인을 받기 전에 시장이다.,
의학 장치 분류에서 유럽

유럽위원회
에 대한 규정 의학 장치에서는 유럽 연합(EU)설립을 통해 의료기기 지침으로 유럽위원회(EC).
유럽에서 시장으로가는 길은 CE 마킹을 얻는 것입니다.
의료 기기를 표시하는 CE 를 얻는 데 필요한 것이 무엇인지 파악하려면 먼저 의료 기기의 EU 분류를 결정해야합니다., 유럽 연합의 의료 기기 규정(EU MDR)에는 기기 클래스를 결정하는 데 필요한 정보가 포함되어 있습니다.
EU MDR2017/745 정 지시어 2001/83/EC,Regulation(EC)No178/2002 및 규제(EC)No1223/2009 및 폐지 위원회 지침 90/385/EEC 및 93/42/EEC. EU MDR 은 2020 년 5 월부터 의료 기기에 대한 필수 규정이 될 것입니다.
을 확인해야 할 경우에 귀하의 의료 장치입니다:
- 비침범성
- 하는 모든 장치에 침투하지 않습니다 신체 구멍을 통해 또는 표면의 몸입니다., 이러한 장치는 일반적으로 클래스 I;그러나,특정 규정 및 예외 적용을 하는 그들을 만들 수 있는 Class II 또는 더 높습니다.
- 침
- 어떤 장치에서,전체 또는 부분적으로,침투,신체 내부의 하나를 통해 몸의 구멍을 통해 또는 표면의 몸입니다.
- Active
- 모든 장치의 동작에 따라 에너지원이 다른 것보다는 생성에 의하여 인체에 대한 목적,또는 중력에 의해,그리고 역할을 하는 변경하여 밀도의거나 변환하는 에너지입니다.,
에 대한 각각의 광범위한 범주가 있는 특정 규칙이 적용된 부속서 VIII 의 새로운 의학 장치 규정입니다. 사용 기간과 결합 된 이러한 범주는 분류를 상당히 간단하게 결정합니다.
예를 들면,장치에서 지속적인 사용을 위한 60 분 간주되는 일시적인 시간,60 분 30 일으로 간주 짧은 기간,그리고 30 일 이상 간주 오래-용어입니다.,
마음에 그를 결정하기 위해,EU 분류,장치의 우리가 사용할 수 있는 매몰 카테테르를 들어 사용전에 대해서는 이 설명서 FDA 분류 수 있습니다.
내 의료 기기가”침습적”범주에 적합하다고 판단한다고 가정 해 봅시다.이것은 내 검색을 규칙 5,6,7 및 8 로 좁 힙니다.
I 할 수 있는 좁은 다음 규칙 아래는 더 나가 알고 있기 때문에 내 의학 장치는 단기 때문에 그것이 사용되는 기간은 24 시간 이상 less than30 일입니다.
거기에서 규칙 7 이 내 장치의 분류에 가장 적용 가능하다는 것을 확인할 수 있습니다.,

출처:EU MDR2017/745Annex VIII5.3
이후 매몰 카테터를 관리하는 의약품,나는 것을 확인할 수 있습니다 내 의학 장치로 간주됩 Class IIb 의학 장치에서는 유럽 시장입니다.
을 결정하는 경로를 시장에서 유럽
유럽연합이 유사한 제품 분류 시스템으로 미국,:
- 클래스 I
- 클래스 IIa
- Class IIb
- Class III
모든 경우에서 의학을 위한 기기에서 판매되는 유럽연합 기술 문서 필요한 단계입니다 프로세스에서 획득 CE Marking. 모든 의료기기 클래스가 유럽에서 필요한 작업으로 통지 신체를 제외하고,그는 클래스가 될 수 있습 자체 인증을 받았습니다.
회사는 또한 유럽에서 제품 등록을 돌볼 수있는 공인 대리인과 협력해야합니다., Article11 최고 질문에 EC 권한을 담당자를 제공하는 데 도움이 될 수 있습니다 추가 설명을 달성하는 방법에 대한 의료기기 등급 분류에서 유럽입니다.
의학 장치 분류에서 캐나다
![]()
캐나다 보건부
의학 장치 규정은 캐나다에서 설립된 캐나다 정부의 규제를 받으며 건강이다.
미국과 EU 와 마찬가지로 캐나다 시장에 판매하려면 먼저 캐나다의 규정에 따라 의료 기기 분류를 결정해야합니다.,
와 유사한 요구 사항에 설명된 EU MDR,건강 캐나다 제공하는 매우 간단하고 쉽게 따라 지도에서 위험을 기반으로 분류 시스템을 위한 비 체외 진단 장치에 대 한 의료 장치 제조업체를 사용하여 판매할 경우 이 시장이다.,
캐나다 보건부의 정의는 네 개의 그룹의 비 체외 진단 의료기기:
- 침 장치(규칙 1-3)
- 비침범성 장치(규칙 4-7 일)
- Active 장치(규칙 8-12 일)
- 특별한 규칙(규칙 13-16)
에 대한 각각의 넓은 범주에있는 규칙의 적용됩니다. 이러한 규칙은 제조업체가 장치의 위험 분류를 결정하기 위해 따라야하는 것입니다.
캐나다에서 이러한 장치를 마케팅 할 경우 경피 카테터 예를 한 번 더 사용하겠습니다.,
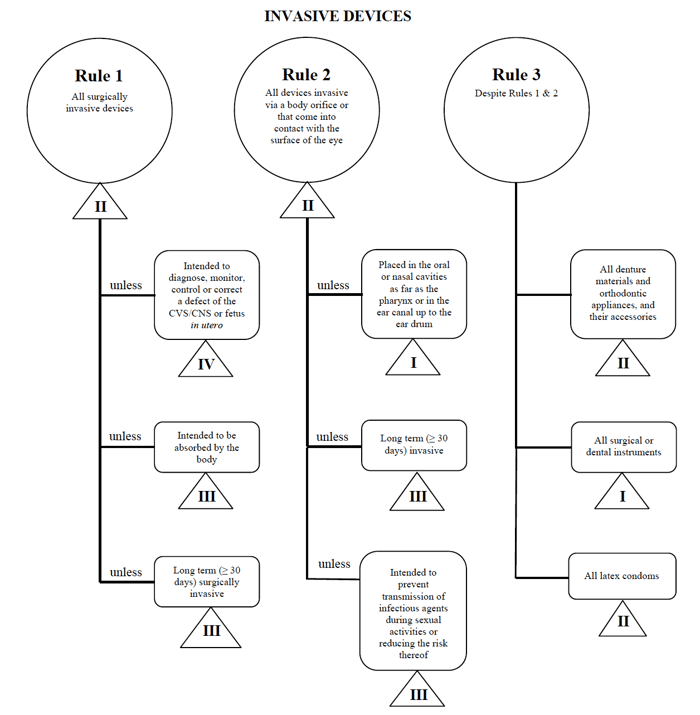
원천:인도에서 위험을 기반으로 분류 시스템을 위한 비 체외 진단 장치
I 하는 의료기기에 맞는”침”카테고리,판매법인과 사무소 및 연구소 내으로 검색하는 규칙 1,2,3.
옵션을 검토 한 후 규칙 1 이 적용되는지 확인합니다.
내 의도 된 용도에 따라,내 의료 기기는 캐나다에서 클래스 II 로 간주됩니다.,
을 결정하는 경로를 시장에서 캐나다
있는 네 가지 수준의 의료 장치를 분류에서 캐나다:
- 클래스 I
- 클래스 II
- Class III
- Class IV
이전에 시장에서 캐나다의 적용을 위한 의학 장치 license. 클래스 i 의료 기기는 라이센스가 필요하지 않습니다. 제조업체는이 과정을 안내합니다 건강 캐나다 지침 문서를 참조 할 수 있습니다.,
Class III 및 Class IV 의료 기기 제조업체는 캐나다 시장 진출을 위해 toc 또는 Health Canada 형식으로 사전 시장 신청서를 제출하여 라이센스를받을 수 있습니다.
또한 MDSAP 로 ISO13485 인증을 획득해야합니다.
업데이트하는 캐나다 보건 규정:süd product services 는
로 2019,모든 의료 장치 제조업체는 판매하는 Class II 의료기기와 높은 캐나다 시장해야 할 부분의 의료 장치는 단일 감사 프로그램(süd product services 는).,
이러한 제조업체를 받아야 통과 전체 감사의 품질 관리 시스템(QMS)프로그램을 통해 캐나다에서. 현재 캐나다,미국,일본,브라질 및 호주를 포함하여 MDSAP 에 참여하는 전 세계 6 개 지역이 있습니다.
Health Canada 가 프로그램에 참여하도록 요구하는 제조업체 이외에도 mdsap 에 참여하는 것은 제조업체에게 선택 사항입니다.,
을 얻을 제공 certification,의료 장치 제조업체에 완료해야 합니다 다음과 같은 세 가지 단계는 다음과 같습니다.
- 응용 프로그램을 검토
- Off-site 문서 감사
- On-site audit
장치 제조업체는 판매하는 캐나다로 받게 될 것입을 연 리뷰와 함께,재인증 감사는 모든입니다. 규정 준수와 의학 장치는 단일 감사 프로그램에 따라 규정에서 ISO13485 표준에 대한 품질 관리 시스템에 대한 의학 장치입니다.,
응용 프로그램을 간소화하기 위해 해당 MDSAP 요구 사항에 따라 제품 및 품질 및 규제 팀을 교육하는 것이 좋습니다.
우리의 무료 갭 평가 도구에 대한 제공 및 ISO13485 는 데 도움이 장치 제조업체를 평가합 QMS 가이드라인을 따라 ISO 과 함께 표준의 요구사항 캐나다 보건부 감사는 조직이(아오)및 다른 지역에 참여하는 프로그램입니다.,이제 전 세계 3 대 장터에서 의료 기기 분류 및 시장 진출 경로를 결정하는 데 필요한 정보와 자원을 보유하고 있습니다.
기기의 분류에 관계없이 각 시장에서 적용되는 규제 지침을 따르는 것이 필수적입니다. 규정 준수를 충족시키는 것은 궁극적으로 장치 및 회사 전체의 운명을 결정하는 품질 관리의 핵심 측면입니다.,
Greenlight Guru 에서 우리는 의료 기기 품질 관리(mdqms)의 중요성을 중요하게 생각합니다. 우리의 MDQMS 소프트웨어는 의료 기기를 위해 특별히 설계된 세계 유일의 솔루션입니다. 그것이 정렬되어 최신 업계 표준의 모범 사례 관리를 위한 제품 디자인 컨트롤 및 위험뿐만 아니라,변화 활동을 제어 및 다른 품질을 이벤트를 제공하고,전체 추적 라이프 사이클 전반에 걸쳐 귀하의 의료 장치입니다.
고 디자인 제어 솔루션에 도움을 가지고 안전한 의료 기기 시장에 더 빨리 더 적은 위험한가?, Greenlight Guru 의 의료 기기 QMS 소프트웨어→
를 빠르게 둘러 보려면 여기를 클릭하십시오.