
Was ich mit Ihnen teilen werde, ist eine Anleitung zur Klassifizierung medizinischer Geräte.
In diesem Handbuch werde ich Ihnen einen schrittweisen Ansatz zur Verfügung stellen, um zu bestimmen, wie Ihr Medizinprodukt von der US-amerikanischen FDA, der Europäischen Kommission und Health Canada klassifiziert wird. Ein grundlegendes Verständnis der regulatorischen Produktklassifizierung ist von unschätzbarem Wert für Ihre Bemühungen, neue Produkte auf den Markt zu bringen.,
Regulatory Classification 101
Die Regeln, die für Ihr Medizinprodukt gelten, hängen davon ab, wie Ihr Produkt von den Regulierungsbehörden klassifiziert wird. Jede Regulierungsbehörde hat verschiedene Klassifikationen für Medizinprodukte definiert.
Die Klassifikationen hängen größtenteils oder in der Regel mit dem wahrgenommenen Risiko des Produkttyp zusammen.
Hersteller von Medizinprodukten, die international verkaufen, müssen sich mit den geltenden Vorschriften dieser Märkte vertraut machen. Dies ist einfacher gesagt als getan und kann für die meisten Hersteller eine Herausforderung sein., Die USA haben ihre Regeln, während Kanada sich an eine andere hält und Europa noch eine.
Glücklicherweise gibt es viele Parallelen zwischen internationalen Vorschriften und Standards für Medizinprodukte. Dieser Leitfaden soll Ihnen zeigen, wie Sie Ihr Gerät in verschiedenen Märkten auf der ganzen Welt klassifizieren können.

Warum spielt die regulatorische Klassifizierung überhaupt eine Rolle?
Aus folgenden Gründen ist es wichtig zu wissen, wie Ihr Medizinprodukt klassifiziert wird:
- Die Produktklassifizierung bestimmt, was Sie tun müssen, bevor Sie Ihr Produkt verkaufen können.,
- Die Produktklassifizierung hilft Ihnen dabei, Anforderungen während der Produktentwicklungsphase festzulegen, insbesondere Designkontrollen.
- Die Produktklassifizierung ist eine wichtige Komponente bei der Bestimmung, wie viel es kostet, Ihr Gerät auf den Markt zu bringen, und gibt Ihnen eine Vorstellung davon, wie lange es dauern wird.
Aus diesem Grund werde ich Ihnen ein wenig Anleitung geben, um besser zu verstehen, was zu tun ist und wie es zu tun ist.,
Beachten Sie, dass die Informationen, die ich bereitstellen werde, dazu beitragen sollen, Sie über die regulatorische Klassifizierung von Medizinprodukten und die für Ihr Medizinprodukt erforderlichen Informationen aufzuklären.
Der folgende Inhalt ist kein umfassender Leitfaden für regulatorische Einreichungen, sollte Ihnen jedoch einige grundlegende Anleitungen und Anweisungen zur Ermittlung des Weges zum Markt geben.
Ich werde mich an die „Big 3“ halten, die Sie wissen sollten, wenn es um die Klassifizierung von Medizinprodukten geht:
- U. S., Food & Drug Administration, Center for Devices & Radiological Health (FDA CDRH)
- europäischen Kommission
- Health Canada
Medizinische Geräte Regulatorische Klassifizierung in den USA

US-amerikanische FOOD & DRUG ADMINISTRATION (FDA)
In den Vereinigten Staaten, medizinische Geräte reguliert durch die Lebensmittel & Drug Administration oder FDA., Die spezifische Niederlassung innerhalb der FDA ist das Zentrum für Geräte & Radiological Health (CDRH).
Die Mission von CDRH ist es, die öffentliche Gesundheit zu schützen und zu fördern. Mit anderen Worten, stellen Sie sicher, dass medizinische Geräte sicher sind. In den USA sind Medizinprodukte entweder Klasse I, Klasse II oder Klasse III. Die FDA-CDRH-Klassifizierung basiert hauptsächlich auf dem Risiko, das das Medizinprodukt darstellt.
Medizinische Geräte der Klasse I gelten im Allgemeinen als geringes Risiko, und medizinische Geräte der Klasse III gelten als das höchste Risiko. Welche Arten von Kontrollen erforderlich sind, hängt von der Klassifizierung Ihres Produkts ab.,
Die Klassifizierung steht in direktem Zusammenhang mit dem Verwendungszweck und den Anwendungshinweisen. Die Unterscheidung zwischen diesen Begriffen ist etwas verwirrend.
- Verwendungszweck ist der allgemeine Zweck des Medizinprodukts oder dessen Funktion (was Sie „behaupten“, dass das Medizinprodukt dies tut).
- Anwendungshinweise beschreiben die Krankheit oder den Zustand, die das medizinische Gerät diagnostiziert, behandelt, verhindert, heilt oder mildert, einschließlich einer Beschreibung der Zielpatientenpopulation.
Denken Sie daran., Die beabsichtigte Verwendung und Indikationen für die Verwendung Ihres Medizinprodukts drücken den Grund aus, warum Sie diese Idee für ein neues Medizinprodukt hatten.
So finden Sie die geltenden FDA-Vorschriften für Ihr Medizinprodukt
Sobald Sie den Verwendungszweck und die Anwendungshinweise definiert haben, müssen Sie jetzt die möglichen Vorschriften und Produktcodes finden. Das Aufspüren der regulatorischen Klassifizierung für Ihr Produkt über die FDA dauert etwas Zeit und Ausdauer.,
Ohne Sie mit zu vielen Details zu langweilen, hat die FDA mehrere allgemeine Kategorien festgelegt, die auf der medizinischen Spezialität in CFR – Titel 21-Lebensmittel und Medikamente basieren: Teile 862 bis 892.
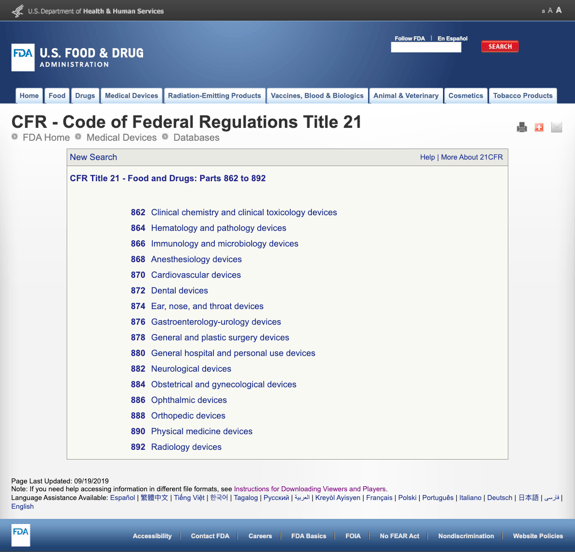
Wenn Sie die möglichen Kategorien finden und auf die FDA-Regulierungsnummer klicken, scheint die Liste der Möglichkeiten plötzlich endlos zu sein. Hier ist eine Teilansicht der Optionen für Teil 870 Herz-Kreislauf-Geräte:

Dies kann frustrierend und überwältigend.,
Wenn Sie eine Regelung finden, die eine mögliche Anpassung zu sein scheint, können Sie auf den Link klicken und weitere Details erhalten, um eine Bestimmung vorzunehmen.
Wenn ich zum Beispiel denke, dass mein Gerät in den Perkutankatheter „870.1250“ passt, klicke ich auf den Link und erhalte folgende Informationen:

Die angegebenen Details geben mir eine Vorstellung davon, ob mein Verwendungszweck und meine Anwendungshinweise mit dieser spezifischen Regelung übereinstimmen. Ich entdecke auch die FDA Device Classification.,
In diesem Beispiel erfahre ich, dass es sich bei meinem Produkt um ein Medizinprodukt der Klasse II (Leistungsstandards) handelt, was bedeutet, dass ich vor der Marktfreigabe einen 510(k) bei der FDA einreichen muss. Ich teile mehr über Arten von FDA Einreichungen weiter in diesem Handbuch.
Weiter-Finden Sie die Produktcodes für Ihr Gerät
Finden Sie die anwendbare Regelung für Sie Medizinprodukt und Klassifizierung ist der erste Teil. Jetzt müssen Sie die entsprechenden Produktcodes finden. So geht ‚ s:
Rufen Sie die FDA-Produktklassifizierungsdatenbank auf und geben Sie die gefundene Regulierungsnummer ein., Wenn Sie mehr als eine Möglichkeit finden, müssen Sie diesen Vorgang für jeden wiederholen.
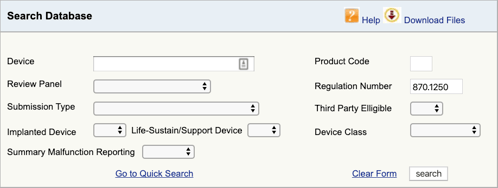
Wenn Sie auf“ Suchen “ klicken, erhalten Sie eine Liste möglicher Produktcodes.
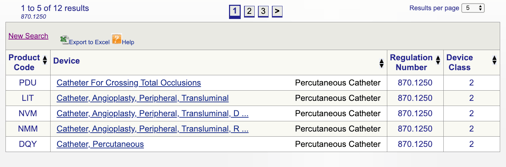
Sie können dann jeden einzelnen Code überprüfen, um die beste Option für Ihr Produkt zu bestimmen, indem Sie auf jeden Code klicken.
Bestimmen Sie Ihren Weg zum Markt IN DEN USA
Die Kenntnis der geltenden Vorschriften und des Produktcodes (wie oben beschrieben) ist erforderlich, damit Sie die Klassifizierung Ihres Medizinprodukts bestimmen können., Sobald Sie diese Informationen haben, können Sie nun den „Pfad“ bestimmen, um Ihr Produkt bei der FDA zu registrieren.,
Die FDA definiert drei regulatorische Kontrollen für jede Medizinproduktklasse:
- Medizinprodukt der Klasse I (geringes bis mittleres Risiko): Allgemeine Kontrollen
- Medizinprodukt der Klasse II (mäßiges bis hohes Risiko): Allgemeine Kontrollen und Sonderkontrollen
- Medizinprodukt der Klasse III (hohes Risiko): Allgemeine Kontrollen und Marktzulassung (PMA)
Lassen Sie es mich auf folgendes herunterkochen:
Wenn Sie feststellen, dass Ihr Produkt „ausgenommen“ ist, dann gelten nur allgemeine Kontrollen und es ist keine formelle FDA-Einreichung erforderlich. Sie müssen jedoch Ihre Einrichtung bei der FDA registrieren und dann das Produkt auflisten.,
Wenn Sie feststellen, dass Ihr Produkt spezielle Kontrollen erfordert, müssen Sie eine 510(k) – Einreichung bei der FDA vorbereiten und die Freigabe erhalten, bevor Sie auf den Markt gehen. Danach müssen Sie Ihre Einrichtung registrieren und das Produkt auflisten.
Wenn Sie feststellen, dass Ihr Produkt eine Vorkaufsgenehmigung erfordert, müssen Sie den FDA-PMA-Prozess befolgen, um die Genehmigung zu erhalten, bevor Sie auf den Markt gehen.,
Medizinprodukteinstufung in Europa

Europäische Kommission
Die Vorschriften für ein Medizinprodukt in der Europäischen Union (EU) werden durch die Medizinprodukterichtlinien der Europäischen Kommission (EG) festgelegt.
Der Weg zum Markt in Europa besteht darin, eine CE-Kennzeichnung zu erhalten.
Um herauszufinden, was erforderlich ist, um eine CE-Kennzeichnung Ihres Medizinprodukts zu erhalten, müssen Sie zunächst die EU-Klassifizierung Ihres Medizinprodukts bestimmen., Die Medizinprodukteverordnung der Europäischen Union (EU MDR) enthält die erforderlichen Informationen zur Bestimmung Ihrer Geräteklasse.
Der EU-MDR 2017/745 ändert die Richtlinie 2001/83/EG, die Verordnung (EG) Nr. 178/2002 und die Verordnung (EG) Nr. 1223/2009 und hebt die Richtlinien 90/385/EWG und 93/42 / EWG des Rates auf. Die EU-MDR wird ab Mai 2020 zur verbindlichen Verordnung für Medizinprodukte.
Sie müssen feststellen, ob Ihr Medizinprodukt:
- Nicht-Invasiv
- Jedes Gerät, das nicht durch eine Öffnung oder die Oberfläche des Körpers in den Körper eindringt., Diese Geräte sind typischerweise Klasse I; Es gelten jedoch bestimmte Regeln und Ausnahmen, die sie zur Klasse II oder höher machen könnten.
- Invasive
- Jedes Gerät, das ganz oder teilweise durch eine Körperöffnung oder durch die Körperoberfläche in den Körper eindringt.
- Aktiv
- Jedes Gerät, dessen Betrieb von einer anderen Energiequelle als der zu diesem Zweck vom menschlichen Körper oder von der Schwerkraft erzeugten abhängt und das durch Ändern der Dichte oder Umwandlung dieser Energie wirkt.,
Für jede der großen Kategorien gelten bestimmte Regeln, die in Anhang VIII der neuen Medizinprodukteverordnung aufgeführt sind. Diese Kategorien in Verbindung mit der Nutzungsdauer machen die Klassifizierung ziemlich einfach.
Beispielsweise wird ein Gerät, das weniger als 60 Minuten im Dauereinsatz ist, als vorübergehende Dauer betrachtet, 60 Minuten bis 30 Tage als kurzfristig und über 30 Tage als langfristig.,
In diesem Sinne können wir zur Bestimmung der EU-Klassifizierung Ihres Geräts das perkutane Katheterbeispiel verwenden, das zuvor in diesem Handbuch für die FDA-Klassifizierung verwendet wurde.
Angenommen, ich stelle fest, dass mein Medizinprodukt in die Kategorie „invasiv“ passt; Dies beschränkt meine Suche auf die Regeln 5, 6, 7 und 8.
Ich kann dann die Regeln weiter eingrenzen, da ich weiß, dass mein Medizinprodukt kurzfristig ist, da es für einen Zeitraum von mehr als 24 Stunden und weniger als 30 Tagen verwendet wird.
Von dort aus kann ich feststellen, dass Regel 7 für die Klassifizierung meines Geräts am besten geeignet ist.,
Quelle: EU MDR 2017/ 745 Anhang VIII 5.3
Da mein Perkutankatheter Medikamente verabreichen wird, kann ich bestätigen, dass mein Medizinprodukt auf dem europäischen Markt als Medizinprodukt der Klasse IIb gilt.
BESTIMMEN SIE IHREN WEG ZUM MARKT IN EUROPA
Die Europäische Union verfügt über ein ähnliches Produktklassifizierungssystem wie die USA.,:
- Class I
- Class IIa
- Class IIb
- Class III
In allen Fällen ist die technische Dokumentation für den Verkauf von Medizinprodukten in der Europäischen Union ein notwendiger Schritt bei der Erlangung der CE-Kennzeichnung. Alle Medizinprodukteklassen in der EU erfordern die Zusammenarbeit mit einer benannten Stelle, mit Ausnahme derjenigen, die Klasse I sind und selbst zertifiziert werden können.
Unternehmen müssen auch mit einem autorisierten Vertreter zusammenarbeiten, um sich um die Produktregistrierung in Europa zu kümmern., Der Artikel 11 Top-Fragen zu EG-Bevollmächtigten können dazu beitragen, weitere Klarstellungen darüber zu geben, wie die Einstufung von Medizinprodukten in Europa erreicht werden kann.
Medizinprodukteklassifizierung in Kanada
![]()
Health Canada
Die Medizinproduktevorschriften in Kanada werden von der kanadischen Regierung festgelegt und von Health Canada reguliert.
Wie die USA und die EU, in den kanadischen Markt zu verkaufen, müssen Sie zuerst die medizinische Geräteklassifikation nach Kanadas Verordnung bestimmen.,
Ähnlich wie die in der EU MDR beschriebenen Anforderungen bietet Health Canada eine ziemlich einfache und leicht zu befolgende Anleitung zum risikobasierten Klassifizierungssystem für Nicht-In-Vitro-Diagnosegeräte, die Hersteller von Medizinprodukten beim Verkauf auf diesem Markt verwenden können.,
Health Canada definiert vier Gruppen von nicht-in-vitro-diagnostischen Medizinprodukten:
- Invasive Geräte (Regeln 1 – 3)
- Nicht-invasive Geräte (Regeln 4 – 7)
- Aktive Geräte (Regeln 8 – 12)
- Sonderregeln (Regeln 13 – 16)
Für jede der großen Kategorien gibt es eine Reihe von Regeln, die gelten. Diese Regeln sollten Hersteller befolgen, um die Risikoklassifizierung ihres Geräts zu bestimmen.
Ich werde das perkutane Katheterbeispiel noch einmal verwenden, wenn ein solches Gerät in Kanada vermarktet wird.,
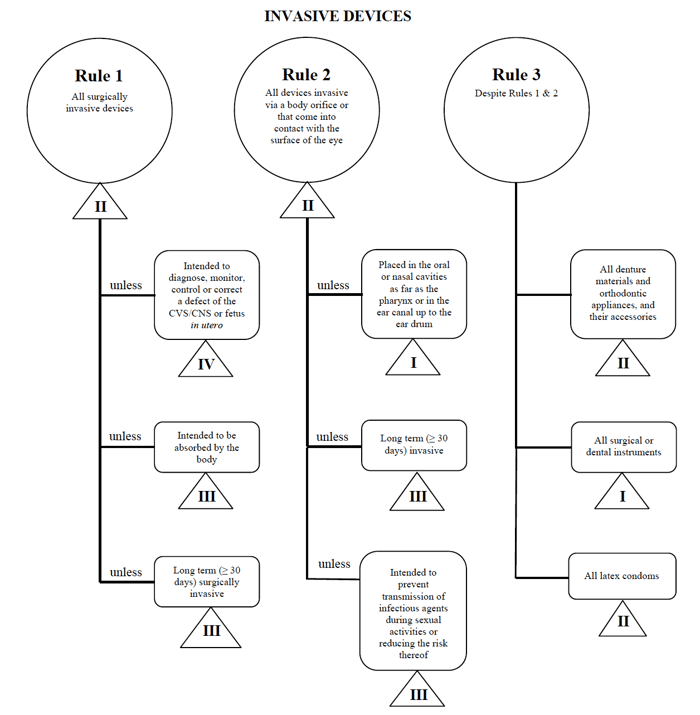
Source: Guidance on the Risk-based Classification System for Non-In-Vitro Diagnostic Devices
ich bestimme meine medizinische Gerät passt in die „invasive“ – Kategorie, Verengung meiner Suche nach unten zu Regeln 1, 2 und 3.
Nach Überprüfung der Optionen stelle ich fest, dass Regel 1 gilt.
Aufgrund meiner beabsichtigten Verwendung gilt mein Medizinprodukt in Kanada als Klasse II.,
BESTIMMEN SIE IHREN WEG ZUM MARKT IN KANADA
In Kanada gibt es vier Stufen von Klassifikationen für Medizinprodukte:
- Class I
- Class II
- Class III
- Class IV
Bevor Sie in Kanada auf den Markt gehen, müssen Sie zuerst eine Lizenz für Medizinprodukte beantragen. Medizinische Geräte der Klasse I benötigen keine Lizenz. Hersteller können auf das Health Canada Guidance Document verweisen, das Sie durch diesen Prozess führt.,
Hersteller von Medizinprodukten der Klassen III und IV können ihre Lizenz erhalten, indem sie einen Premarket-Antrag im ToC-oder Health Canada-Format für den Eintritt in den kanadischen Markt einreichen.
Sie müssen auch die ISO 13485-Zertifizierung mit MDSAP erhalten.
Aktualisierung der Health Canada-Vorschriften: MDSAP
Ab Januar 2019 müssen alle Hersteller von Medizinprodukten, die Medizinprodukte der Klasse II und höher auf dem kanadischen Markt verkaufen, Teil des Medical Device Single Audit Program (MDSAP) sein.,
Diese Hersteller müssen ein vollständiges Audit ihres Qualitätsmanagementsystems (QMS) durch das Programm in Kanada durchlaufen und bestehen. Derzeit gibt es 6 Regionen auf der ganzen Welt, die an MDSAP teilnehmen, darunter Kanada, USA, Japan, Brasilien und Australien.
Abgesehen von den Herstellern, die Health Canada für die Teilnahme am Programm benötigt, ist die Teilnahme an MDSAP für Hersteller optional.,
Um eine MDSAP-Zertifizierung zu erhalten, müssen Medizinproduktehersteller die folgenden drei Schritte ausführen:
- Anwendung und Überprüfung
- Off-site documentation Audit
- On-site Audit
Gerätehersteller, die nach Kanada verkaufen, werden jährlich überprüft, wobei jedes dritte Jahr ein Rezertifizierungsaudit durchgeführt wird. Die Einhaltung des einzigen Auditprogramms für Medizinprodukte basiert auf der Einhaltung der Richtlinien der Norm ISO 13485 für Qualitätsmanagementsysteme für Medizinprodukte.,
Wir empfehlen, Ihre Produkt -, Qualitäts-und Regulierungsteams in den geltenden MDSAP-Anforderungen zu schulen, um Ihre Anwendung zu optimieren.
Unser kostenloses Gap Assessment Tool für MDSAP und ISO 13485 hilft Geräteherstellern, die QMS-Richtlinien der ISO-Norm neben den Anforderungen von Health Canada Auditing Organizations (AO) und anderen am Programm teilnehmenden Regionen zu bewerten.,
Final Thoughts
Sie haben jetzt die Informationen und Ressourcen, die Sie benötigen, um Ihre Medizinproduktklassifikation und Ihren Marktpfad auf den drei größten Marktplätzen der Welt zu bestimmen.
Unabhängig von der Klassifizierung Ihres Geräts müssen Sie unbedingt die gesetzlichen Richtlinien befolgen, die in jedem Markt gelten. Die Einhaltung der Compliance ist ein Schlüsselaspekt des Qualitätsmanagements, der letztendlich über das Schicksal Ihres Geräts und Ihres Unternehmens als Ganzes entscheidet.,
Bei Greenlight Guru legen wir Wert auf die Bedeutung des Qualitätsmanagements für Medizinprodukte (MDQMS). Unsere MDQMS-Software ist die weltweit einzige Lösung, die speziell für Medizinprodukte entwickelt wurde. Es ist auf die neuesten Best Practices des Industriestandards für die Verwaltung von Produktdesignkontrollen und-risiken sowie Änderungskontrollaktivitäten und anderen Qualitätsereignissen abgestimmt und bietet vollständige Rückverfolgbarkeit während des gesamten Lebenszyklus Ihres Medizinprodukts.
Suchen Sie nach einer Design-Steuerungslösung, mit der Sie sicherere Medizinprodukte mit weniger Risiko schneller auf den Markt bringen können?, Klicken Sie hier, um eine kurze Tour von Greenlight Gurus Medical Device QMS Software zu machen →