
wat ik met u ga delen is een gids voor de wettelijke classificatie van medische hulpmiddelen.
in deze gids zal ik u een stapsgewijze aanpak geven om te bepalen hoe uw medisch hulpmiddel zal worden geclassificeerd door de Amerikaanse FDA, de Europese Commissie en Health Canada. Het krijgen van een basiskennis van regelgevende productclassificatie zal van onschatbare waarde zijn voor uw inspanningen om nieuwe producten op de markt te brengen.,
wettelijke classificatie 101
de regels die van toepassing zijn op uw medisch hulpmiddel hangen af van de manier waarop uw product is geclassificeerd door de regelgevende instanties. Elk regelgevend agentschap heeft verschillende classificaties voor medische hulpmiddelen gedefinieerd.
de classificaties houden grotendeels of in de regel verband met het waargenomen risico van de productsoort.
fabrikanten van medische hulpmiddelen die internationaal verkopen, moeten zich vertrouwd maken met de toepasselijke regelgeving van die markten. Dit is makkelijker gezegd dan gedaan en kan een uitdaging voor de meeste fabrikanten., De VS heeft zijn set van regels, terwijl Canada zich aan een andere houdt, en Europa nog steeds een andere.
gelukkig zijn er veel parallellen tussen internationale regelgeving en normen voor medische hulpmiddelen. Deze gids is ontworpen om u te laten zien hoe u uw apparaat in verschillende markten over de hele wereld te classificeren.

waarom doet de wettelijke classificatie er zelfs toe?
weten hoe uw medisch hulpmiddel geclassificeerd is, is om de volgende redenen belangrijk:
- productclassificatie bepaalt wat u moet doen voordat u uw product kunt verkopen.,
- productclassificatie zal u helpen eisen vast te stellen tijdens de productontwikkelingsfase, met name ontwerpcontroles.
- productclassificatie is een belangrijk onderdeel om te bepalen hoeveel het kost om uw apparaat op de markt te brengen en u een idee te geven van hoe lang het zal duren.
hierdoor zal ik je een beetje begeleiding geven om beter te begrijpen wat je moet doen en hoe je het moet doen.,
opmerking: de informatie die ik u ga verstrekken is bedoeld om u te informeren over de wettelijke classificatie van medische hulpmiddelen en wat er nodig is voor uw medische hulpmiddelen.
de volgende inhoud is geen uitgebreide gids voor regulatory submissions, maar zou u enige basisbegeleiding en richting moeten geven bij het bepalen hoe u het pad naar de markt kunt bepalen.
Ik houd het bij de “big 3” die u moet weten als het gaat om classificatie van medische hulpmiddelen:
- U. S., Eten & Drug Administration Center for Devices & Radiological Health (FDA CDRH)
- Europese Commissie
- Health Canada
Medische Apparaat Indeling volgens de Regelgeving in de verenigde staten

AMERIKAANSE FOOD & DRUG ADMINISTRATION (FDA)
In de Verenigde Staten, medische apparatuur wordt geregeld door het Voedsel & Drug Administration of FDA., De specifieke tak binnen de FDA is het Centrum voor apparaten & Radiologische Gezondheid (CDRH).
CDRH heeft tot taak de volksgezondheid te beschermen en te bevorderen. Met andere woorden, zorg ervoor dat medische hulpmiddelen veilig zijn. In de VS zijn medische hulpmiddelen Klasse I, Klasse II of klasse III. de FDA CDRH-classificatie is voornamelijk gebaseerd op het risico dat het medische hulpmiddel vormt.
medische hulpmiddelen van klasse I worden over het algemeen als laag risico beschouwd en medische hulpmiddelen van klasse III worden als het hoogste risico beschouwd. De soorten controles vereist is afhankelijk van de classificatie van uw product.,
De indeling houdt rechtstreeks verband met het beoogde gebruik en de indicaties voor gebruik. Het onderscheid tussen deze termen is een beetje verwarrend.
- het beoogde gebruik is het algemene doel van het medische hulpmiddel of de functie ervan (wat u beweert dat het medische hulpmiddel doet).
- indicaties voor gebruik beschrijf de ziekte of aandoening die het medische hulpmiddel zal diagnosticeren, behandelen, voorkomen, genezen of verzachten, met inbegrip van een beschrijving van de doelpatiëntpopulatie.
houd dit in gedachten., Het beoogde gebruik en de indicaties voor het gebruik van uw medisch hulpmiddel geven de reden aan waarom u dit idee voor een nieuw medisch hulpmiddel had.
hoe vindt u de toepasselijke FDA-voorschriften voor uw medisch hulpmiddel
zodra u het beoogde gebruik en de indicaties voor gebruik hebt gedefinieerd, moet u nu de mogelijke voorschriften en productcodes vinden. Het opsporen van wettelijke Classificatie voor uw product via de FDA neemt een beetje tijd en doorzettingsvermogen.,
zonder u te vervelen met al te veel details, heeft de FDA verschillende algemene categorieën vastgesteld op basis van de medische specialiteit in CFR Titel 21 – Food and Drugs: delen 862 tot 892.
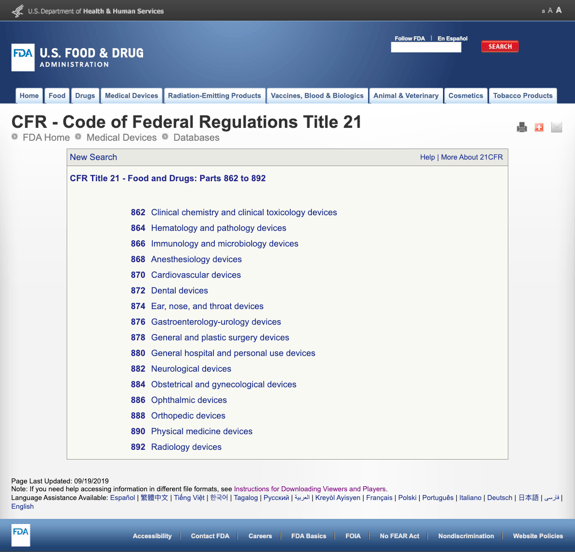
wanneer u de mogelijke categorieën vindt en op het reglementnummer van de FDA klikt, lijkt de lijst met mogelijkheden plotseling eindeloos. Hier is een gedeeltelijke weergave van de opties voor deel 870 cardiovasculaire apparaten:

Dit kan frustrerend en overweldigend zijn.,
wanneer u een verordening vindt die mogelijk past, kunt u op de link klikken en meer details krijgen om een bepaling te maken.
bijvoorbeeld, als ik denk dat mijn apparaat past in “870.1250 percutane katheter,” Klik ik op de link en krijg deze informatie:

de verstrekte gegevens geven me een idee of mijn beoogde gebruik en indicaties voor gebruik overeenkomen met deze specifieke verordening. Ik ontdek ook de FDA apparaat classificatie.,
in dit voorbeeld leer ik dat mijn product Een klasse II medisch hulpmiddel is (prestatienormen), wat betekent dat ik een 510(k) moet indienen bij de FDA voordat ik marktklaring krijg. Ik deel meer over soorten FDA inzendingen verder in deze gids.
volgende-Zoek de productcodes die van toepassing zijn op uw apparaat
het vinden van de toepasselijke regelgeving voor uw medisch hulpmiddel en classificatie is het eerste deel. Nu moet u de toepasselijke productcodes vinden. Hier is hoe:
Ga naar de FDA-Productclassificatiedatabase en typ het regelnummer dat u hebt gevonden., Als u meer dan één mogelijkheid vindt, moet u dit proces voor elk herhalen.
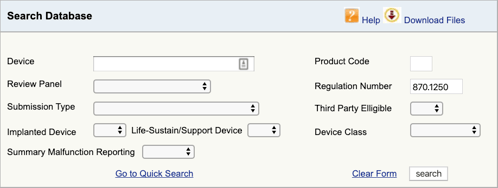
wanneer u op” search ” klikt, krijgt u een lijst met mogelijke productcodes.
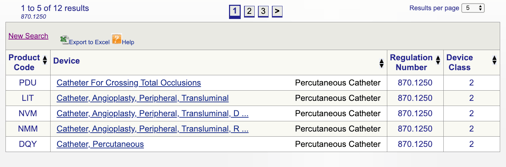
u kunt vervolgens elke afzonderlijke code bekijken om de beste optie voor uw product te bepalen door op elke code te klikken.
het bepalen van uw weg naar de markt in de VS
Het kennen van de toepasselijke regelgeving en productcode (zoals hierboven beschreven) is noodzakelijk om de classificatie van uw medisch hulpmiddel te bepalen., Zodra u deze informatie hebt, kunt u nu het “pad” bepalen om uw product geregistreerd te krijgen bij de FDA.,
FDA definieert drie regelgevende controles voor elk medisch hulpmiddel klasse:
- medisch hulpmiddel Klasse I (laag tot matig risico): Algemene Controles
- Klasse II medical device (matig tot hoog risico): General Controls en Speciale Gereedschappen
- Klasse III medische apparaat (hoog risico): General Controls en Premarket Approval (PMA)
Laat ik kook het op neer:
Als u uw product is “vrijgesteld” en vervolgens alleen algemene controles van toepassing zijn en geen formele FDA indiening vereist. U hoeft, echter, moet uw vestiging registreren bij de FDA en vervolgens een lijst van het product.,
Als u merkt dat uw product speciale controles vereist, betekent dit dat u een 510(k) – aanvraag bij de FDA moet voorbereiden en goedkeuring moet ontvangen voordat u naar de markt gaat. Daarna, je nodig hebt om uw vestiging te registreren en een lijst van het product.
Als u merkt dat uw product premarket goedkeuring vereist, betekent dit dat u het FDA PMA proces moet volgen om goedkeuring te ontvangen voordat u naar de markt gaat.,
classificatie van medische hulpmiddelen in Europa

Europese Commissie
de voorschriften voor een medisch hulpmiddel in de Europese Unie (EU) zijn vastgesteld door de richtlijnen voor medische hulpmiddelen door de Europese Commissie (EC).
de weg naar de markt in Europa is het verkrijgen van een CE-markering.
om erachter te komen wat nodig is om een CE-markering op uw medisch hulpmiddel te verkrijgen, moet u eerst de EU-classificatie van uw medisch hulpmiddel bepalen., De verordening medische hulpmiddelen van de Europese Unie (EU MDR) bevat de nodige informatie om uw apparaatklasse te bepalen.
EU MDR 2017/745 wijzigt Richtlijn 2001/83/EG, Verordening (EG) nr. 178/2002 en Verordening (EG) nr. 1223/2009 en tot intrekking van de richtlijnen 90/385/EEG en 93/42 / EEG van de Raad. EU-MDR wordt vanaf mei 2020 de verplichte verordening voor medische hulpmiddelen.
u moet bepalen of uw medisch hulpmiddel:
- niet-invasief
- elk hulpmiddel is dat niet door een opening of het oppervlak van het lichaam in het lichaam dringt., Deze apparaten zijn meestal Klasse I; er zijn echter bepaalde regels en uitzonderingen van toepassing die ze klasse II of hoger kunnen maken.
- invasief
- elk hulpmiddel dat geheel of gedeeltelijk in het lichaam binnendringt, hetzij door een lichaamsopening, hetzij door het oppervlak van het lichaam.
- actief
- elk apparaat waarvan de werking afhankelijk is van een andere energiebron dan die welke voor dat doel door het menselijk lichaam wordt opgewekt, of door zwaartekracht, en dat werkt door de dichtheid van die energie te veranderen of om te zetten.,
voor elk van de brede categorieën gelden bepaalde regels die zijn uiteengezet in bijlage VIII van de nieuwe verordening inzake medische hulpmiddelen. Deze categorieën in combinatie met de gebruiksduur maken het bepalen van de indeling vrij eenvoudig.
een hulpmiddel dat gedurende minder dan 60 minuten continu wordt gebruikt, wordt bijvoorbeeld beschouwd als een voorbijgaande duur, 60 minuten tot 30 dagen als een kortdurend gebruik en langer dan 30 dagen als een langdurig gebruik.,
om de EU-classificatie van uw apparaat te bepalen, kunnen we het voorbeeld van de percutane katheter gebruiken dat eerder in deze gids voor FDA-classificatie werd gebruikt.
stel dat ik bepaal dat mijn medisch hulpmiddel past in de categorie” invasief”; dit beperkt mijn zoekopdracht tot Regels 5, 6, 7 en 8.
Ik kan de regels verder beperken, omdat ik weet dat mijn medisch hulpmiddel van korte duur is omdat het langer dan 24 uur en minder dan 30 dagen wordt gebruikt.
vanaf daar kan Ik bepalen dat regel 7 het meest van toepassing is op de classificatie van mijn apparaat.,

bron: EU MDR 2017/ 745 bijlage VIII 5.3
aangezien mijn percutane katheter geneesmiddelen zal toedienen, kan ik bevestigen dat mijn medisch hulpmiddel op de Europese markt wordt beschouwd als een medisch hulpmiddel van klasse IIb.
bepalen van uw weg naar de markt in Europa
De Europese Unie heeft een soortgelijk productclassificatiesysteem als de VS.,:
- Klasse I
- klasse IIa
- klasse IIb
- klasse III
in alle gevallen voor medische hulpmiddelen die in de Europese Unie worden verkocht, is technische documentatie een vereiste stap in het proces voor het verkrijgen van de CE-markering. Alle klassen van medische hulpmiddelen in de EU vereisen samenwerking met een aangemelde instantie, met uitzondering van die welke klasse I zijn en zelf gecertificeerd kunnen worden.
bedrijven zullen ook moeten samenwerken met een gemachtigde vertegenwoordiger om de productregistratie in Europa te verzorgen., De belangrijkste vragen in artikel 11 over door de EG gemachtigde vertegenwoordigers kunnen helpen om meer duidelijkheid te verschaffen over de manier waarop in Europa de classificatie van medische hulpmiddelen kan worden bereikt.
classificatie van medische hulpmiddelen in Canada
![]()
Health Canada
de voorschriften voor medische hulpmiddelen in Canada worden vastgesteld door de regering van Canada en gereguleerd door Health Canada.
net als de VS en de EU, moet u eerst de classificatie van medische hulpmiddelen onder de Canadese regelgeving bepalen om op de Canadese markt te verkopen.,
net als de eisen in de EU MDR biedt Health Canada een vrij eenvoudige en gemakkelijk te volgen leidraad voor het risicogebaseerde classificatiesysteem voor niet-In-vitrodiagnostiek die fabrikanten van medische hulpmiddelen kunnen gebruiken bij verkoop op deze markt.,
Health Canada definieert vier groepen medische hulpmiddelen voor niet-in-vitrodiagnostiek:
- invasieve hulpmiddelen (regels 1 – 3)
- niet-invasieve hulpmiddelen (regels 4 – 7)
- actieve hulpmiddelen (regels 8 – 12)
- bijzondere regels (regels 13 – 16)
voor elk van de grote categorieën zijn er een aantal regels van toepassing. Deze regels zijn wat fabrikanten moeten volgen om de risico-indeling van hun hulpmiddel te bepalen.
Ik zal het percutane katheter voorbeeld nogmaals gebruiken in het geval van het op de markt brengen van een dergelijk apparaat in Canada.,
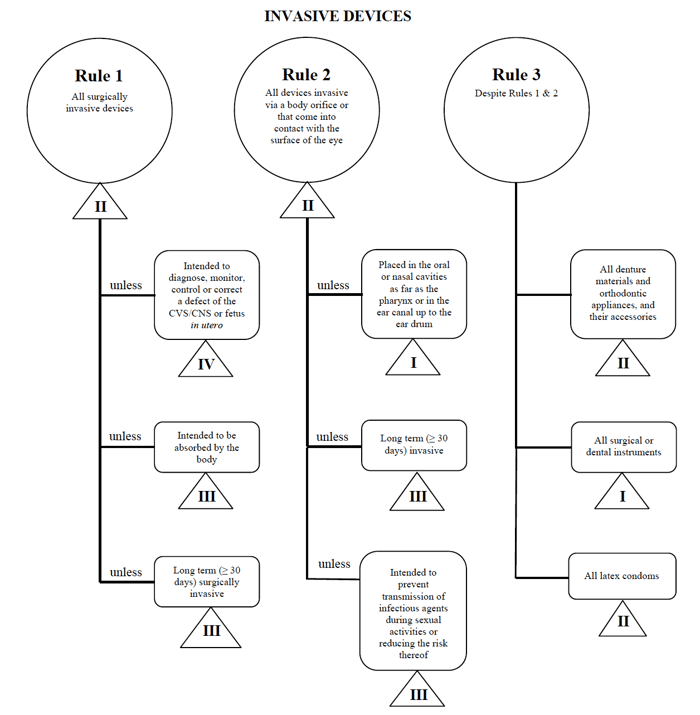
Source: Guidance on the Risk based Classification System for Non-In Vitro Diagnostic Devices
i determinate my medical device fits into the “invasive” category, narrow my search down to Rules 1, 2, and 3.
na het bekijken van de opties, bepaal ik dat Regel 1 van toepassing is.
Op basis van mijn beoogde gebruik wordt mijn medisch hulpmiddel in Canada beschouwd als klasse II.,
bepalen van uw weg naar de markt in CANADA
Er zijn vier niveaus van classificaties van medische hulpmiddelen in Canada:
- Klasse I
- klasse II
- klasse III
- klasse IV
voordat u naar de markt in Canada gaat, moet u eerst een vergunning voor medische hulpmiddelen aanvragen. Voor medische hulpmiddelen van klasse I is geen vergunning vereist. Fabrikanten kunnen verwijzen naar de Health Canada guidance document, die u door dit proces loopt.,
fabrikanten van medische hulpmiddelen van klasse III en klasse IV kunnen hun licentie ontvangen door een premarket-aanvraag in te dienen, in de ToC-of Health Canada-formaten, om de Canadese markt te betreden.
u moet ook ISO 13485-certificering verkrijgen met MDSAP.
Update to Health Canada Regulations: MDSAP
vanaf januari 2019 moeten alle fabrikanten van medische hulpmiddelen die medische hulpmiddelen van klasse II en hoger op de Canadese markt verkopen, deel uitmaken van het Single Audit Programme (Mdsap) voor medische hulpmiddelen.,
deze fabrikanten moeten een volledige audit van hun kwaliteitsmanagementsysteem (QMS) via het programma in Canada ondergaan en doorstaan. Er zijn momenteel 6 regio ‘ s over de hele wereld die deelnemen aan MDSAP, waaronder Canada, de VS, Japan, Brazilië en Australië.
behalve de fabrikanten die door Health Canada verplicht zijn aan het programma deel te nemen, is deelname aan MDSAP facultatief voor fabrikanten.,
om mdsap-certificering te verkrijgen, moeten fabrikanten van medische hulpmiddelen de volgende drie stappen uitvoeren:
- Application and review
- Off-site documentation audit
- On-site audit
fabrikanten van hulpmiddelen die in Canada verkopen, zullen jaarlijks worden onderworpen aan evaluaties, met een hercertificeringsaudit om de drie jaar. De naleving van het Single Audit programma voor medische hulpmiddelen is gebaseerd op het voldoen aan de richtlijnen van de ISO 13485-norm voor kwaliteitsmanagementsystemen voor medische hulpmiddelen.,
Wij raden u aan uw product-en kwaliteits-en regelgevende teams te trainen in de toepasselijke mdsap-vereisten om uw toepassing te stroomlijnen.
onze free gap assessment tool voor MDSAP en ISO 13485 helpt apparaatmakers de QMS-richtlijnen te beoordelen vanuit de ISO-Standaard, naast de vereisten van Health Canada auditing organizations (AO) en andere regio ‘ s die deelnemen aan het programma.,
Final Thoughts
u hebt nu de informatie en middelen die u nodig hebt om uw classificatie van medische hulpmiddelen te bepalen en het pad naar de markt in de drie grootste markten ter wereld.
ongeacht de classificatie van uw apparaat, is het noodzakelijk dat u de wettelijke richtlijnen volgt die van toepassing zijn in elke markt. Voldoen aan compliance is een belangrijk aspect van kwaliteitsmanagement dat uiteindelijk zal beslissen over het lot van uw apparaat en bedrijf als geheel.,
bij Greenlight Guru waarderen we het belang van medical device quality management (mdqms). Onze mdqms-software is de enige oplossing ter wereld die speciaal is ontworpen voor medische hulpmiddelen. Het is afgestemd op de nieuwste industriestandaard best practices voor het beheer van productontwerpcontroles en risico ‘ s, evenals change control-activiteiten en andere kwaliteitsgebeurtenissen, waardoor volledige traceerbaarheid gedurende de levenscyclus van uw medische apparatuur wordt geboden.
Op zoek naar een ontwerpbesturingsoplossing om u te helpen veiliger medische hulpmiddelen sneller op de markt te brengen met minder risico?, Klik hier voor een snelle rondleiding door de QMS-software van Greenlight goeroe voor medische hulpmiddelen →