
Hva jeg skal dele med deg er en guide til medisinsk enhet forskrifter klassifisering.
I denne håndboken, jeg vil gi deg en steg-for-steg tilnærming for å bestemme hvordan din medisinske utstyret vil bli klassifisert av AMERIKANSKE FDA, Eu-Kommisjonen, og Health Canada. Å få en grunnleggende forståelse av regulatoriske produkt klassifisering vil være uvurderlig for din innsats for å bringe nye produkter til markedet.,
Forskrifter Klassifisering 101
De regler som gjelder for din medisinske enheten er avhengig av hvordan produktet er klassifisert av tilsynsorganer. Hver reguleringsmyndighet har definert flere ulike klassifiseringer for medisinsk utstyr.
klassifikasjonene er, for det meste, eller som en generell regel, knyttet til det som oppfattes som risiko for produktet.
Medisinsk utstyr produsenter som selger internasjonalt må gjøre seg kjent med gjeldende bestemmelser i disse markedene. Dette er lettere sagt enn gjort, og kan være en utfordring for de fleste produsenter., USA har sine regler, mens Canada fester seg til en annen, og Europa annen fortsatt.
Heldigvis, det er mange paralleller mellom internasjonale medisinske utstyret forskrifter og standarder. Denne guiden er laget for å vise deg hvordan å klassifisere enheten i ulike markeder rundt om i verden.

Hvorfor Gjør Forskrifter Klassifisering Selv Saken?
å Vite hvordan det medisinske utstyret er klassifisert saker for følgende grunner:
- Produkt klassifisering vil finne ut hva du må gjøre før du kan selge ditt produkt.,
- Produkt klassifisering vil hjelpe deg å etablere krav i produktutviklingsfasen, spesielt design kontroller.
- Produkt klassifisering er en viktig komponent i å bestemme hvor mye det vil koste å få enheten til å markedsføre og gi deg en viss idé om hvor lang tid det vil ta.
på Grunn av dette, jeg kommer til å gi deg en liten bit av veiledning til å bedre forstå hva du skal gjøre og hvordan du gjør det.,
Merk, den informasjonen jeg er i ferd med å gi er ment å bidra til å utdanne deg på medisinsk enhet forskrifter klassifisering og hva som kreves for medisinsk enhet.
følgende innhold er ikke en omfattende guide til pålagte innleveringer, men burde gi deg noen grunnleggende veiledning og retning på å identifisere hvordan å etablere vei til markedet.
jeg vil holde seg til «big 3» du bør vite når det kommer til medisinsk enhet klassifisering:
- USA, Mat & Drug Administration, Center for Devices & Radiological Health (FDA CDRH)
- Eu-Kommisjonen
- Health Canada
Medisinsk Enhet Forskrifter Klassifisering i USA

US FOOD & DRUG ADMINISTRATION (FDA)
I Usa, medisinsk utstyr er regulert av Mat & Drug Administration, eller FDA., Det bestemt gren innen FDA er Center for Devices & Radiological Health (CDRH).
oppgave CDRH er å beskytte og fremme folkehelsen. Med andre ord, sikre at medisinsk utstyr som er trygge. I USA, medisinsk utstyr er enten Klasse i, Klasse II og Klasse III. FDA CDRH-klassifisering er i hovedsak basert på risiko det medisinske utstyret positurer.
Klasse i medisinsk utstyr er generelt ansett som lav risiko og Klasse III medisinsk utstyr som er sett på som den høyeste risikoen. Hvilke typer kontroller som kreves er avhengig av produktets klassifisering.,
Klassifisering er direkte relatert til tiltenkt bruk og indikasjoner for bruk. Skillet mellom disse vilkårene er litt forvirrende.
- Tiltenkt Bruk er de generelle formålet med den medisinske enheten eller dens funksjon (hva du «krav» det medisinske utstyret gjør).
- Indikasjoner for Bruk beskrive den sykdom eller tilstand som det medisinske utstyret vil diagnostisere, behandle, forebygge, helbrede, eller redusere, inkludert en beskrivelse av målet pasienten befolkningen.
ha dette i tankene., Tiltenkt bruk og indikasjoner for bruk av medisinsk utstyr express grunnen til at du hadde denne ideen for et nytt medisinsk utstyr.
Hvordan for Å Finne den Gjeldende FDA-Forskrifter for Medisinsk Enhet
Når du definerer tiltenkt bruk og indikasjoner for bruk, nå må du finne mulig forskrifter og produkt-koder. Sporing ned forskrifter klassifisering for ditt produkt via FDA tar en liten bit av tid og utholdenhet.,
Uten kjedelig du med for mange detaljer, FDA har etablert flere generelle kategorier basert på medisinsk spesialitet i CFR Tittel 21 – Mat og Medisiner: Deler 862 til 892.
Når du finner den mulige kategorier og klikk på FDA regulering tall, liste av muligheter plutselig virker endeløse. Her er en delvis utsikt over alternativer for Del 870 Kretsløpssystem enheter:

Dette kan være frustrerende og overveldende.,
Når du finner en regulering som synes å være en mulig tilpasning, kan du klikke på linken og få mer informasjon til å gjøre en beslutning.
For eksempel, hvis jeg tror at min enhet passer i «870.1250 Perkutan kateteret,» jeg klikker på linken og få denne informasjonen:

detaljer forutsatt gi meg noen anelse om min tiltenkt bruk og indikasjoner for bruk i samsvar med dette bestemt regulering. Jeg har også oppdage FDA enheten klassifisering.,
I dette eksempelet har jeg vite at mitt produkt er et Klasse II medisinsk enhet (performance standards), som betyr at jeg må sende inn en 510(k) til FDA før å få markedet klaring. Jeg deler mer om typer av FDA bidrag videre i denne guiden.
Neste – Finn Produkt Koder som gjelder for enheten
Finne den gjeldende forskriften for deg medisinsk enhet og klassifisering er den første delen. Nå må du finne det aktuelle produktet koder. Her er hvordan:
Gå til FDA Produkt Klassifisering Database og skriv i forskriften nummeret du har funnet., Hvis du finner mer enn én mulighet, så må du gjenta denne prosessen for hver.
Når du klikker «søk», vil du få en liste over mulige produkt koder.
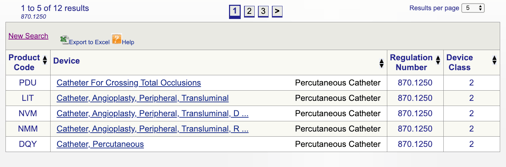
Du kan så se over hver enkelt kode for å finne ut det beste alternativet for ditt produkt ved å klikke på hver enkelt kode.
å Bestemme Din Vei til Markedet I USA
å Vite gjeldende regler og produkt-kode (som beskrevet ovenfor) er nødvendig for deg å bestemme klassifiseringen av det medisinske utstyret., Når du har denne informasjonen, vil du nå være i stand til å finne «veien» for å få produktet som er registrert med FDA.,
FDA definerer tre pålagte kontroller for hver medisinsk utstyr klasse:
- Klasse i medisinsk enhet (lav til moderat risiko): Generelle Kontroller
- Klasse II medisinsk enhet (moderat til høy risiko): Generelle Kontroller og Spesielle Kontroller
- Klasse III medisinsk enhet (høy risiko): Generelle Kontroller og Premarket Godkjenning (PMA)
La meg koker det ned til dette:
Hvis du finner produktet er «fritatt», er bare general styrer gjelder, og ingen formell FDA innsending er nødvendig. Du må imidlertid registrere din etablissement med FDA og deretter liste produktet.,
Hvis du finner produktet krever spesiell kontroll, dette betyr at du er nødt til å forberede en 510(k) bidrag til FDA og får klarering før du går til markedet. Etter at du trenger å registrere deg for etablering og listen produktet.
Hvis du finner produktet krever premarket godkjenning, dette betyr at du er nødt til å følge FDA PMA prosessen med å få godkjenning før du går til markedet.,
Medisinsk Enhet Klassifisering i Europa

Eu-Kommisjonen
regelverket for medisinsk utstyr i Europeiske Union (EU) er etablert gjennom den Medisinske Enheten for Direktiver fra Eu-Kommisjonen (EC).
veien til markedet i Europa er å oppnå en CE-merking.
for Å finne ut hva som er nødvendig for å få et CE-merking av medisinsk enhet, må du først bestemme EU-klassifisering av det medisinske utstyret., Den Europeiske Union er medisinsk enhet forordning (EU MDR) omfatter den informasjon som er nødvendig for å avgjøre enheten klasse.
EU MDR 2017/745 endrer Direktiv 2001/83/EF, Forordning (EF) Nr 178/2002 og Forordning (EF) Nr 1223/2009 og om oppheving av Rådsdirektiv 90/385/EØF og 93/42/EØF. EU-MDR vil bli obligatorisk forskrift for medisinsk utstyr som starter i Mai 2020.
vil Du trenger for å finne ut om din medisinske utstyret er:
- Ikke-Invasive
- en hvilken som Helst enhet som ikke trenge inn i kroppen gjennom en munnstykket eller overflaten av kroppen., Disse enhetene er vanligvis Klasse i, men visse regler og unntak gjelder som kunne gjøre dem i Klasse II eller høyere.
- Invasive
- Alle enheter som, helt eller delvis, trenger inn i kroppen, enten gjennom en kropp munnstykket eller gjennom overflaten av kroppen.
- Aktiv
- en hvilken som Helst enhet som drift er avhengig av en energikilde annet enn det som er generert av den menneskelige kroppen for det formålet, eller ved hjelp av gravitasjon, og som fungerer ved å endre tettheten av eller konvertering som energi.,
For hver av de kategorier, er det visse regler som gjelder, som er skissert i Vedlegg VIII av ny medisinsk enhet regulering. Disse kategoriene sammen med varigheten for bruk gjøre å bestemme klassifisering ganske grei.
For eksempel, en enhet i kontinuerlig bruk for under 60 minutter anses forbigående varighet 60 minutter til 30 dager, regnes som kortsiktig, og over 30 dager anses som langsiktige.,
Med det i tankene, for å avgjøre EU-klassifisering av enheten, kan vi bruke perkutan kateteret eksempel brukt tidligere i denne håndboken for FDA-klassifisering.
La oss si at jeg bestemme min medisinske enheten passer inn i det «fremmede» – kategorien; dette begrenser søket min ned til Reglene 5, 6, 7, og 8.
da kan jeg begrense regler ned ytterligere siden jeg vet at min medisinsk enhet er kort sikt fordi det er brukt for en periode som er større enn 24 timer og mindre enn 30 dager.
Fra det, er jeg i stand til å bestemme at Regelen 7 er mest aktuelt å klassifisering av min enhet.,

Kilde: EU-MDR 2017/ 745 Vedlegg VIII 5.3
Siden min perkutan kateteret vil administrere medisinene, jeg kan bekrefte at min medisinske implantatet regnes som en Klasse IIb medisinsk enhet i den Europeiske markedsplassen.
å BESTEMME DIN VEI TIL MARKEDET I EUROPA
Den Europeiske Union har et liknende produkt klassifisering system som det AMERIKANSKE,:
- Klasse i
- Klasse IIa
- Klasse IIb
- Klasse III
I alle tilfeller for medisinsk utstyr som skal selges i Eu, teknisk dokumentasjon er et nødvendig trinn i prosessen med å få CE-Merking. Alle medisinsk enhet klasser i EU krever at du arbeider med et teknisk kontrollorgan, med unntak for de som er i Klasse i og kan være selv-sertifisert.
Selskaper vil også behovet for å arbeide med en Autorisert Representant til å ta vare på produktet registrering i Europa., Artikkel 11 Toppen Spørsmål om EF Autoriserte Representanter kan bidra til å gi en nærmere avklaring på hvordan man skal oppnå medisinsk enhet klassifisering i Europa.
Medisinsk Enhet Klassifisering i Canada
![]()
Health Canada
Det medisinske enheter bestemmelser i Canada er etablert av Regjeringen i Canada og regulert av Health Canada.
Som USA og EU, for å selge inn det Kanadiske markedet, må du først finne ut av det medisinske utstyret for klassifisering under Canadas regulering.,
Ligner de kravene som er beskrevet i EU-MDR, Health Canada, gir et ganske grei og enkel å følge Veiledning på en risikobasert Klassifisering System for Non-In Vitro-Diagnostisk Utstyr for medisinsk utstyr produsenter å bruke når du selger inn i dette markedet.,
Helse Canada definerer fire grupper av ikke-in vitro-diagnostisk medisinsk utstyr:
- Invasive Enheter (Regler 1 – 3)
- Ikke-Invasive Enheter (Reglene 4 – 7)
- Aktive Enheter (Regler 8 – 12)
- Spesielle Regler (13 – 16)
For hver av de kategorier, det er et sett med regler som gjelder. Disse reglene er hva produsentene bør følge for å bestemme risiko klassifisering av sin enhet.
jeg vil bruke perkutan kateteret eksempel en gang i tilfelle av markedsføring slik enhet i Canada.,
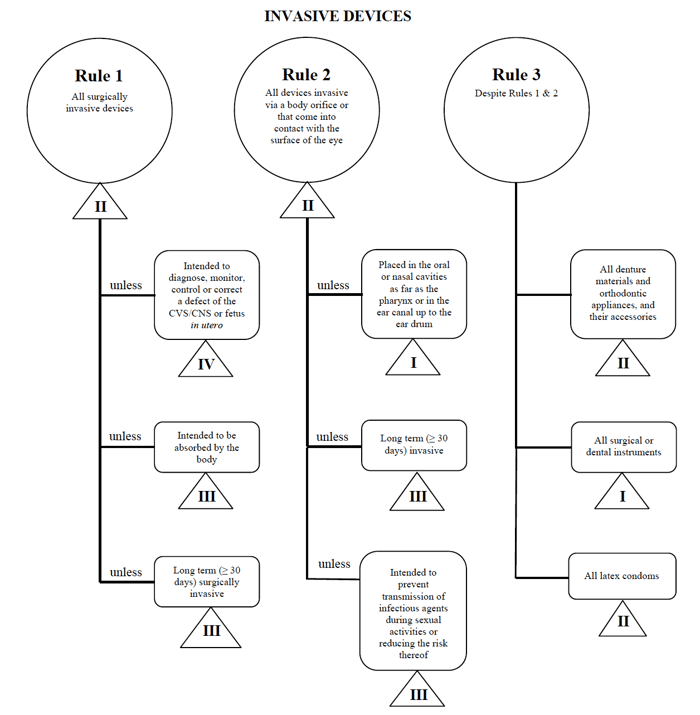
Kilde: Veiledning om risikobasert Klassifisering System for Non-In Vitro-Diagnostisk Utstyr
jeg ut min medisinske enheten passer inn i det «fremmede» – kategorien, innsnevring mitt søk ned til Regler 1, 2, og 3.
Etter at du har vurdert alternativene, jeg bestemmer som Regel 1 gjelder.
Basert på mine hadde til hensikt å bruke, min medisinske utstyret er ansett som Klasse II i Canada.,
å BESTEMME DIN VEI TIL MARKEDET I CANADA
Det er fire nivåer av medisinsk enhet klassifikasjoner i Canada:
- Klasse i
- Klasse II
- Klasse III
- Klasse IV
Før du går til markedet i Canada, må du først søke etter en medisinsk enhet lisens. Klasse i medisinsk utstyr som ikke krever en lisens. Produsenter kan referere til Helse Canada styringsdokument, som leder deg gjennom denne prosessen.,
Produsenter av Klasse III og Klasse IV medisinske enheter kan motta sin lisens, ved å sende en premarket program, enten Innholdsfortegnelsen eller Helse Canada formater, for å skrive inn det Canadiske markedet.
vil Du også trenger å få tak i ISO 13485-sertifisering med MDSAP.
Oppdater til Helse Canada Forskrifter: MDSAP
Som av januar 2019, alt medisinsk utstyr produsenter som selger medisinsk Klasse II-utstyr og høyere til den Kanadiske markedet må være en del av det Medisinske utstyret for Enkelt revisjonsprogram (MDSAP).,
Disse produsentene må gjennomgå og bestå en full revisjon av sine quality management system (QMS) gjennom programmet i Canada. Det er for tiden 6 regioner rundt om i verden som deltar i MDSAP, inkludert Canada, USA, Japan, Brasil og Australia.
Bortsett fra de produsentene som kreves av Health Canada for å delta i programmet, deltakelse i MDSAP er frivillig for produsenter.,
for Å få MDSAP sertifisering av medisinsk utstyr produsenter må fullføre følgende tre trinn:
- Program og omtale
- Off-site dokumentasjon revisjon
- På-stedet-revisjon
Enheten beslutningstakere selger i Canada vil være gjenstand for årlige vurderinger, med en resertifisering revisjon hvert tredje år. Samsvar med den Medisinske Enheten for Enkelt revisjonsprogram er basert på å møte retningslinjer fra ISO 13485 standard på quality management systemer for medisinsk utstyr.,
Vi anbefaler trening for dine produkt og kvalitet og regulatoriske lagene i gjeldende MDSAP krav til å effektivisere din søknad.
Vår gratis gap assessment tool for MDSAP og ISO 13485 hjelper enheten beslutningstakere vurdere QMS retningslinjer fra ISO-standarden sammen med kravene til Helse Canada revisjon organisasjoner (AO) og andre regioner som deltar i programmet.,
Siste Tanker
Du har nå informasjon og ressurser du trenger for å bestemme din medisinske utstyret for klassifisering og veien til markedet i tre av de største markedsplasser rundt om i verden.
Uavhengig av klassifisering av enheten, er det viktig at du følger lovpålagte retningslinjer som gjelder i hvert enkelt marked. Møte compliance er en sentral del av kvalitetsstyring som til slutt vil avgjøre skjebnen til enheten og selskapet som helhet.,
På Greenlight Guru, vi verdien og viktigheten av medisinske utstyret for quality management (MDQMS). Våre MDQMS programvare er den eneste løsningen i verden som er utformet spesielt for medisinsk utstyr. Det er på linje med den nyeste industristandard beste praksis for håndtering av produktet, design kontroller og risiko, samt endre kontroll-aktiviteter og andre kvalitet hendelser, som gir full sporbarhet gjennom hele livssyklusen av det medisinske utstyret.
Leter etter et design-kontroll løsning for å hjelpe deg bringe tryggere medisinsk utstyr som er på markedet med mindre risiko?, Klikk her for å ta en rask tur Greenlight Guru ‘ s Medisinske utstyret QMS programvare →