
私があなたと共有しようとしているのは、医療機器規制分類のガイドです。
このガイドでは、米国FDA、欧州委員会、およびカナダ保健省によって医療機器がどのように分類されるかを決定するためのステップバイステップのアプローチについて説明します。 規制製品の分類の基本的な理解を得ることは、新製品を市場に投入するための取り組みにとって非常に貴重です。,
規制分類101
医療機器に適用されるルールは、規制当局による製品の分類方法によって異なります。 各規制機関は、医療機器のためのいくつかの異なる分類を定義しています。
分類は、ほとんどの場合、または一般的な原則として、製品タイプの知覚されるリスクに関連しています。
国際的に販売する医療機器メーカーは、これらの市場の適用される規制に精通する必要があります。 これはされるより容易な言われ、ほとんどの製造業者のための挑戦である場合もある。, 米国にはその一連の規則があり、一方カナダは別の規則に従い、ヨーロッパはまだ別の規則に従います。
幸いなことに、国際的な医療機器規制と規格の間には多くの類似点があります。 このガイドは、世界中のさまざまな市場でデバイスを分類する方法を示すように設計されています。

なぜ規制分類が重要なのですか?
医療機器がどのように分類されているかを知ることは、次の理由により重要です。
- 製品分類は、製品を販売する前に何をすべきかを決定します。,
- 製品分類は、製品開発段階、特に設計コントロールの間に要件を確立するのに役立ちます。
- 製品分類は、デバイスを市場に投入するためにどれくらいの費用がかかるかを決定する上で重要な要素です。
このため、私は何をすべきか、そしてそれを行う方法をよりよく理解するためのガイダンスを少し提供するつもりです。,
私が提供しようとしている情報は、医療機器規制の分類とあなたの医療機器に必要なものについてあなたを教育するのに役立つことを意図
以下のコンテンツは、規制当局の提出に関する包括的なガイドではありませんが、市場への道筋を確立する方法を特定するための基本的なガイダンスと指示を提供する必要があります。
私はそれが医療機器の分類に来るときあなたが知っておくべき”ビッグ3″に固執します:
- 米国, 食品&医薬品局、デバイスセンター&放射線健康(FDA CDRH)
- 欧州委員会
- カナダ保健省
米国における医療機器規制分類

米国食品&医薬品局(fda)
米国では、医療機器は食品によって規制されています&医薬品局、またはFda。, FDA内の特定のブランチは、デバイスのセンター&放射線健康(CDRH)です。
CDRHの使命は、公衆衛生を保護し、促進することです。 言い換えれば、医療機器が安全であることを確認します。 米国では、医療機器はクラスI、クラスII、またはクラスIIIのいずれかです。FDA CDRH分類は、主に医療機器がもたらすリスクに基づいています。
クラスIの医療機器は一般に低リスクとみなされ、クラスIIIの医療機器は最高リスクと見なされます。 必要なコントロールの種類は、製品の分類によって異なります。,
分類は、意図された使用および使用の適応症に直接関係している。 これらの用語の区別は少し混乱します。
- 意図された用途とは、医療機器またはその機能の一般的な目的です(医療機器が”主張する”もの)。<li><li>使用の適応症は、標的患者集団の記載を含めて、医療デバイスが診断、治療、予防、治癒、または軽減する疾患または状態を記載する。
これを覚えておいてください。, あなたの医療機器の使用のための意図されていた使用そして徴候は新しい医療機器のためのこの考えをなぜ有したか理由を表現する。
医療機器に適用されるFDA規制を見つける方法
使用目的と使用の適応症を定義したら、可能な規制と製品コードを見つける必要があります。 FDAを介して製品の規制分類を追跡するには、少しの時間と忍耐が必要です。,
あまりにも多くの詳細であなたを退屈させることなく、FDAはCFRタイトル21-食品と医薬品:パート862から892の医療専門に基づいていくつかの一般的なカテゴリーを確立しています。
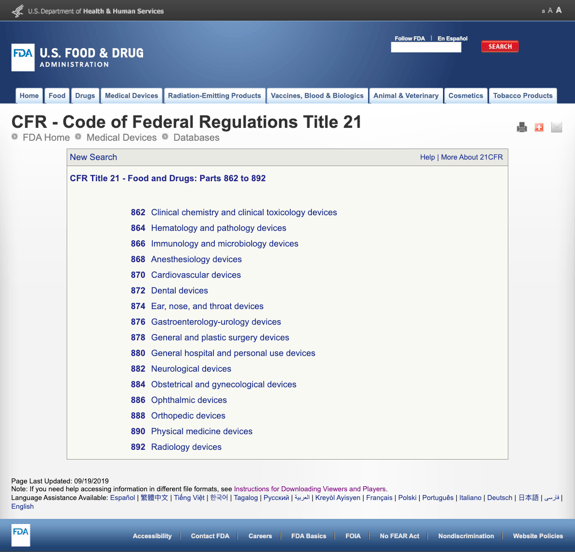
可能なカテゴリを見つけてFDA規制番号をクリックすると、可能性のリストは突然無限のように見えます。 パート870心臓血管デバイスのオプションの部分的なビューは次のとおりです。

これはイライラして圧倒的になる可能性があります。,
あなたが可能な適合であると思われる規制を見つけたら、あなたはリンクをクリックして、決定を行うために、より多くの詳細を取得することが
たとえば、私のデバイスが”870.1250Percutaneous cathutaneousカテーテル”に適合すると思われる場合、リンクをクリックしてこの情報を取得します。

私の意図された使用および使用の適応症がこの特定の規制に合っているかどうか、提供された詳細は私にいくつかのアイデアを与えます。 私はまた、FDAデバイス分類を発見します。,
この例では、私の製品がクラスII医療機器(性能基準)であることを知り、市場クリアランスを取得する前に510(k)をFDAに提出する必要があります。 私はこのガイドでさらにFDAの提出の種類についての詳細を共有しています。
次–お使いのデバイスに適用される製品コードを見つける
お使いの医療機器および分類に適用される規制を見つけることは、最初の部分です。 これで、該当する製品コードを見つける必要があります。
FDA製品分類データベースに移動し、見つかった規制番号を入力します。, 複数の可能性が見つかった場合は、それぞれに対してこのプロセスを繰り返す必要があります。
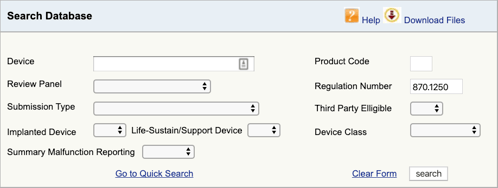
“検索”をクリックすると、可能な製品コードのリストが表示されます。
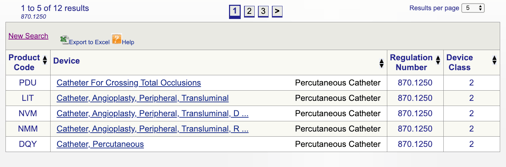
各コードをクリックすることで、個々のコードを確認して製品に最適なオプションを決定できます。
米国での市場投入への道筋を決定する
医療機器の分類を決定するためには、適用される規制および製品コード(上記)を知る必要があります。, この情報を入手すると、製品をFDAに登録するための”パス”を決定できるようになります。,
FDAは、各医療機器クラスについて三つの規制管理を定義しています。
- クラスI医療機器(低リスクから中リスク):一般管理および特別管理
- クラスIII医療機器(高リスク):一般管理および市販前承認(PMA)
これに煮詰めてみましょう。
お客様の製品が”免除されていることがわかった場合、
“その後、一般的な管理のみが適用され、正式なfdaの提出は必要ありません。 ただし、施設をFDAに登録してから、製品をリストアップする必要があります。,
あなたのプロダクトが特別な制御を要求することを見つければ、これはFDAに510(k)服従を準備し、市場に行く前に整理を受け取らなければならないことを意味します。 その後、あなたの施設を登録し、製品をリストする必要があります。
あなたの製品が市販前の承認を必要としている場合、これはあなたが市場に出る前に承認を受けるためにFDA PMAプロセスに従わなければならないことを意味します。,
ヨーロッパにおける医療機器分類

欧州委員会
欧州連合(EU)における医療機器の規制は、欧州委員会(EC)による医療機器指
ヨーロッパでの市場への道は、CEマーキングを取得することです。
医療機器をマーキングするCEを取得するために必要なものを把握するには、まず医療機器のEU分類を決定する必要があります。, 欧州連合の医療機器規則(EU MDR)には、お客様の機器クラスを決定するために必要な情報が含まれています。
EU MDR2017/745は、指令2001/83/EC、規則(EC)No178/2002および規則(EC)No1223/2009を改正し、評議会の指令90/385/EECおよび93/42/EECを廃止します。 EU MDRは2020年から医療機器の必須規制となる予定です。
あなたの医療機器が次のものであるかどうかを判断する必要があります。
- 非侵襲的
- オリフィスまたは身体の表面を通って身体に浸透しない, これらのデバイスは通常クラスIですが、クラスII以上にすることができる特定の規則および例外が適用されます。 侵襲的
- 身体オリフィスを介して、または身体の表面を介して、身体の内部に全体または一部を貫通する任意のデバイスを含む。
- アクティブ
- その目的のために人体によって生成されたもの以外のエネルギー源に依存し、そのエネルギーの密度を変えたり変換したりすること,
- クラスI
- クラスIIa
- クラスIIb
- クラスIII
- Non襲性デバイス規則4-7
- アクティブデバイス規則8-12
- 特別規則規則13-16
- Class I
- Class II
- Class III
- Class IV
幅広いカテゴリーごとに、新しい医療機器規則の附属書VIIIに概説されている特定の規則が適用されます。 これらのカテゴリは、使用期間と相まって、分類の決定をかなり簡単にします。
たとえば、60分以下の連続使用のデバイスは一時的な持続時間とみなされ、60分から30日は短期とみなされ、30日以上は長期とみなされます。,
このことを念頭に置いて、デバイスのEU分類を決定するために、このガイドで以前に使用した経皮的カテーテルの例をFDA分類に使用できます。
私の医療機器が”侵襲的”カテゴリに適合していると判断したとしましょう。これにより、検索はルール5、6、7、および8に絞り込まれます。
私の医療機器は24時間以上30日未満の期間使用されているため、短期間であることがわかっているので、ルールをさらに絞り込むことができます。
そこから、私はルール7が自分のデバイスの分類に最も適用可能であると判断することができます。,

出典:EU MDR2017/745Annex VIII5.3
私の経皮的カテーテルは医薬品を投与するため、私の医療機器が欧州市場でクラスIIb医療機器と見なされていることを確認することができます。
ヨーロッパでの市場投入への道のりを決定する
欧州連合は米国と同様の製品分類システムを持っています,:
欧州連合内で販売される医療機器のすべての場合において、CEマーキングを取得するプロセスにおいて、技術文書が必 EU内のすべての医療機器クラスは、クラスIであり、自己認証可能なものを除き、Notified Bodyとの作業を必要とします。
企業はまた、ヨーロッパでの製品登録の世話をするために権限のある代表者と協力する必要があります。, EC認定代理人に関する第11条のトップ質問は、ヨーロッパで医療機器の分類を達成する方法についてさらに明確にするのに役立ちます。
カナダの医療機器分類
![]()
Health Canada
カナダの医療機器規制は、カナダ政府によって確立され、カナダ保健省によって規制されています。
米国およびEUと同様に、カナダ市場に販売するには、まずカナダの規制に基づく医療機器分類を決定する必要があります。,
EU MDRで概説されている要件と同様に、カナダ保健省は、医療機器メーカーがこの市場に販売する際に使用する非体外診断装置のリスクベースの分類システムに関するガイダンスにかなり簡単で簡単に従うことができます。,侵襲性デバイス規則1-3
幅広いカテゴリーごとに、適用される一連の規則があります。 これらのルールは、メーカーがデバイスのリスク分類を決定するために従うべきものです。
カナダでこのようなデバイスを販売する場合は、経皮的カテーテルの例をもう一度使用します。,
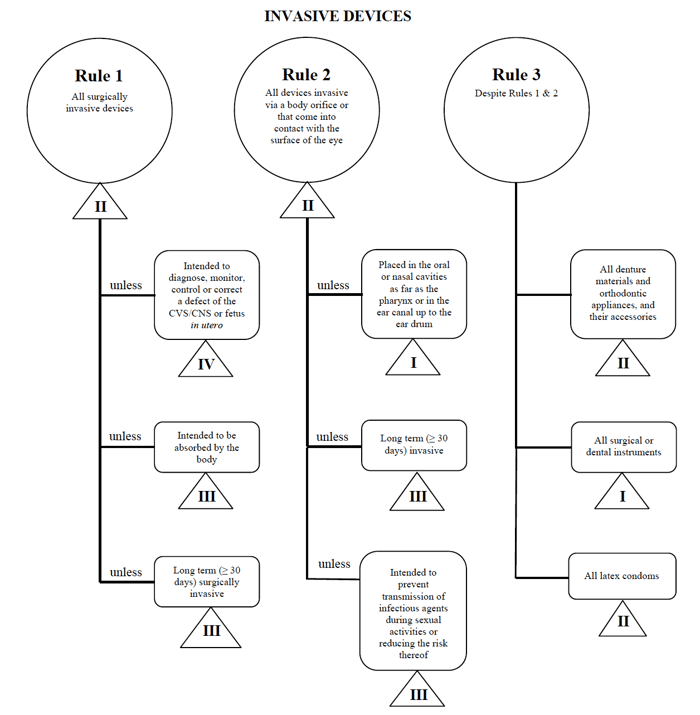
出典:非体外診断装置のリスクベース分類システムに関するガイダンス
私は自分の医療機器が”侵襲的”カテゴリに適合していると判断し、検索をルール1、2、および3に絞り込みます。
オプションを確認した後、ルール1が適用されると判断します。
私の使用目的に基づいて、私の医療機器はカナダではクラスIIとみなされます。,
カナダでの市場への道のりを決定する
カナダでは、医療機器分類には四つのレベルがあります。
カナダで市場に出る前に、まず医療機器ライセンスを申請する必要があります。 クラスI医療機器にはライセンスは必要ありません。 製造業者は、このプロセスを説明するHealth Canadaガイダンス文書を参照することができます。,
クラスIIIおよびクラスIV医療機器の製造業者は、カナダ市場に参入するために、ToCまたはカナダ保健省のいずれかの形式で市販前申請書を提出することにより、ライセンスを受け取ることができます。
また、MDSAPでISO13485認証を取得する必要があります。
カナダ保健省規制への更新:MDSAP
2019年現在、カナダ市場にクラスII以上の医療機器を販売するすべての医療機器メーカーは、医療機器単一監査プログラム(MDSAP)の一部である必要があります。,
これらの製造業者は、カナダのプログラムを通じて品質管理システム(QMS)の完全な監査を受け、合格しなければなりません。 現在、MDSAPに参加している地域は、カナダ、米国、日本、ブラジル、オーストラリアなど、世界中に6つあります。
カナダ保健省がプログラムに参加するために必要とする製造業者とは別に、製造業者にとってMDSAPへの参加は任意である。,
MDSAP認証を取得するには、医療機器メーカーは次の三つのステップを完了する必要があります。
- 申請とレビュー
- オフサイト文書監査
- オンサイト監査
カナダに販売するデバイスメーカーは、毎年審査を受け、三年毎に再認証監査を受けます。 医療機器の単一監査プログラムへの準拠は、医療機器の品質管理システムに関するISO13485規格のガイドラインを満たすことに基づいています。,
お客様のアプリケーションを合理化するために、該当するMDSAP要件で製品および品質および規制チームを訓練することをお勧めします。
MDSAPおよびISO13485のための無料のgap評価ツールは、デバイスメーカーがカナダ保健省監査機関(AO)およびプログラムに参加する他の地域の要件とともに、ISO標準からのQMSガイドラインを評価するのに役立ちます。,
最終的な考え
あなたは今、あなたが世界中の最大の市場の三つの市場であなたの医療機器の分類と市場への道を決定するために必要
お使いのデバイスの分類にかかわらず、各市場で適用される規制ガイドラインに従うことが不可欠です。 コンプライアンスを満たすことは、最終的にお客様のデバイスと会社全体の運命を決定する品質管理の重要な側面です。,
Greenlight Guruでは、医療機器品質管理(MDQMS)の重要性を大切にしています。 当社のMDQMSソフトウェアは、医療機器専用に設計された世界で唯一のソリューションです。 これは、製品設計管理やリスク、変更管理活動やその他の品質イベントを管理するための最新の業界標準のベストプラクティスと整合しており、医療機器のライフサイクル全体にわたって完全なトレーサビリティを提供します。
より安全な医療機器をより少ないリスクでより速く市場に投入するのに役立つ設計制御ソリューションをお探しですか?, Greenlight Guruの医療機器QMSソフトウェアのクイックツアーに参加するにはここをクリック→