
Quello che sto per condividere con voi è una guida per la classificazione di regolamentazione dei dispositivi medici.
In questa guida, ti fornirò un approccio passo-passo per determinare come il tuo dispositivo medico sarà classificato dalla FDA degli Stati Uniti, dalla Commissione europea e da Health Canada. Ottenere una conoscenza di base della classificazione dei prodotti normativi sarà inestimabile per i vostri sforzi per portare nuovi prodotti sul mercato.,
Classificazione normativa 101
Le regole applicabili al dispositivo medico dipendono dalla classificazione del prodotto da parte delle agenzie di regolamentazione. Ogni agenzia di regolamentazione ha definito diverse classificazioni per i dispositivi medici.
Le classificazioni sono, per la maggior parte o come regola generale, correlate al rischio percepito del tipo di prodotto.
I produttori di dispositivi medici che vendono a livello internazionale devono familiarizzare con le normative applicabili di tali mercati. Questo è più facile a dirsi che a farsi e può essere una sfida per la maggior parte dei produttori., Gli Stati Uniti hanno il loro insieme di regole, mentre il Canada aderisce ad un altro, e l’Europa un altro ancora.
Fortunatamente, ci sono molti parallelismi tra le normative e gli standard internazionali sui dispositivi medici. Questa guida è stata progettata per mostrare come classificare il dispositivo in diversi mercati in tutto il mondo.

Perché la classificazione normativa è importante?
Sapere come viene classificato il tuo dispositivo medico è importante per i seguenti motivi:
- La classificazione del prodotto determinerà cosa devi fare prima di poter vendere il tuo prodotto.,
- La classificazione dei prodotti ti aiuterà a stabilire i requisiti durante la fase di sviluppo del prodotto, in particolare i controlli di progettazione.
- La classificazione del prodotto è una componente importante nel determinare quanto costerà portare il tuo dispositivo sul mercato e darti un’idea di quanto tempo ci vorrà.
A causa di questo, ho intenzione di fornire un po ‘ di guida per capire meglio cosa fare e come farlo.,
Nota, le informazioni che sto per fornire hanno lo scopo di aiutarti a istruirti sulla classificazione normativa dei dispositivi medici e su ciò che è richiesto per il tuo dispositivo medico.
Il seguente contenuto non è una guida completa ai contributi normativi, ma dovrebbe fornire alcune indicazioni e indicazioni di base sull’identificazione di come stabilire il percorso verso il mercato.
Mi atterrò al “big 3” che dovresti sapere quando si tratta di classificazione dei dispositivi medici:
- U. S., Food & la Somministrazione del Farmaco, il Centro per i Dispositivi & Salute Radiologica (FDA CDRH)
- la Commissione Europea
- Health Canada
dispositivi Medici di Regolamentazione Classificazione in USA

FOOD & DRUG ADMINISTRATION (FDA)
Negli Stati Uniti, i dispositivi medici sono regolati dalla Food & Drug Administration, o FDA., Il ramo specifico all’interno della FDA è il Centro per i dispositivi & Radiological Health (CDRH).
La missione di CDRH è proteggere e promuovere la salute pubblica. In altre parole, assicurarsi che i dispositivi medici siano sicuri. Negli Stati Uniti, i dispositivi medici sono di classe I, Classe II o classe III. La classificazione FDA CDRH si basa principalmente sul rischio che il dispositivo medico pone.
I dispositivi medici di classe I sono generalmente considerati a basso rischio e i dispositivi medici di Classe III sono considerati il rischio più elevato. I tipi di controlli richiesti dipendono dalla classificazione del prodotto.,
La classificazione è direttamente correlata all’uso previsto e alle indicazioni per l’uso. La distinzione tra questi termini è un po ‘ confusa.
- L’uso previsto è lo scopo generale del dispositivo medico o della sua funzione (ciò che “rivendichi” il dispositivo medico).
- Indicazioni per l’uso descrivere la malattia o la condizione il dispositivo medico diagnosticherà, curerà, preverrà, curerà o attenuerà, inclusa una descrizione della popolazione di pazienti target.
Tenetelo a mente., L’uso previsto e le indicazioni per l’uso del dispositivo medico esprimono il motivo per cui hai avuto questa idea per un nuovo dispositivo medico.
Come trovare le normative FDA applicabili per il tuo dispositivo medico
Una volta definite la destinazione d’uso e le indicazioni per l’uso, ora devi trovare le possibili normative e codici prodotto. Rintracciare la classificazione normativa per il tuo prodotto tramite FDA richiede un po ‘ di tempo e perseveranza.,
Senza annoiarti con troppi dettagli, la FDA ha stabilito diverse categorie generali basate sulla specialità medica nel titolo CFR 21 – Cibo e droghe: Parti da 862 a 892.
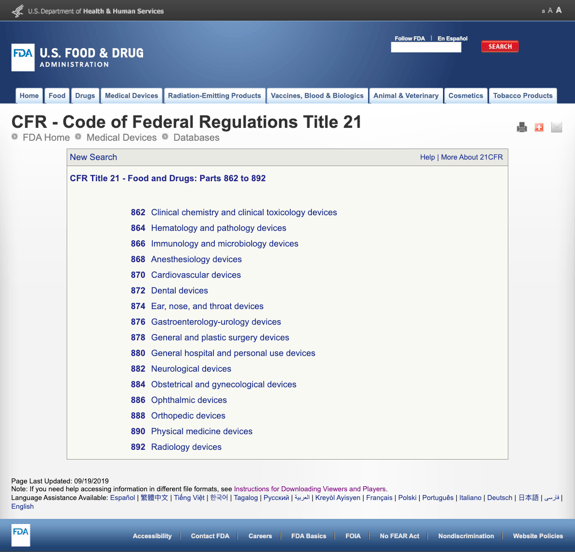
Quando trovi le possibili categorie e fai clic sul numero del regolamento FDA, l’elenco delle possibilità sembra improvvisamente infinito. Ecco una vista parziale delle opzioni per la parte 870 Dispositivi cardiovascolari:

Questo può essere frustrante e travolgente.,
Quando si trova un regolamento che sembra essere una possibile misura, è possibile fare clic sul link e ottenere maggiori dettagli per fare una determinazione.
Per esempio, se penso che il mio dispositivo si inserisce nella “870.1250 Percutaneo di catetere,” clicco il link e ottenere queste informazioni:

I dati forniti darmi qualche idea se la mia destinazione d’uso e indicazioni per l’utilizzo in linea con questo regolamento specifico. Scopro anche la classificazione dei dispositivi FDA.,
In questo esempio, apprendo che il mio prodotto è un dispositivo medico di classe II (standard di prestazioni), il che significa che dovrò presentare un 510(k) alla FDA prima di ottenere l’autorizzazione al mercato. Condivido di più sui tipi di osservazioni FDA più avanti in questa guida.
Avanti-Trova i codici dei prodotti applicabili al tuo dispositivo
Trovare il regolamento applicabile per te dispositivo medico e classificazione è la prima parte. Ora è necessario trovare i codici prodotto applicabili. Ecco come:
Vai al database di classificazione dei prodotti FDA e digita il numero di regolamento che hai trovato., Se trovi più di una possibilità, dovrai ripetere questo processo per ciascuno.
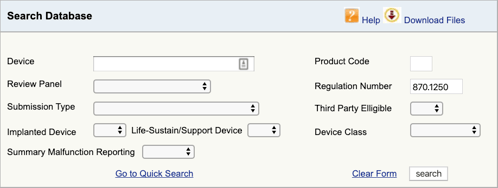
Quando si fa clic su “cerca”, si otterrà un elenco di possibili codici prodotto.
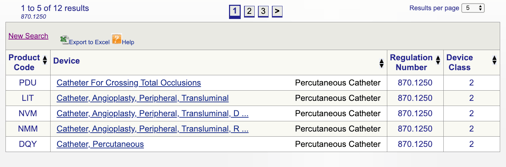
È quindi possibile rivedere ogni singolo codice per determinare l’opzione migliore per il prodotto facendo clic su ciascun codice.
Determinare il percorso verso il mercato NEGLI STATI UNITI
Conoscere il regolamento applicabile e il codice del prodotto (come descritto sopra) è necessario per determinare la classificazione del dispositivo medico., Una volta che hai queste informazioni, ora sarai in grado di determinare il “percorso” per ottenere il tuo prodotto registrato con FDA.,
la FDA definisce tre controlli normativi per ogni dispositivo medico di classe:
- medici di Classe I dispositivo (da basso a moderato rischio): Controlli Generali
- Classe II dispositivo medico (moderato o alto rischio): Controlli Generali e Controlli Speciali
- Classe III dispositivi medici (ad alto rischio): Controlli di carattere Generale e Premarket di Approvazione (PMA)
mi Lasciate bollire fino a questo:
Se si trova il prodotto è “esente”, solo controlli di carattere generale si applicano e non formale FDA presentazione è necessaria. Tuttavia, è necessario registrare il proprio stabilimento con FDA e quindi elencare il prodotto.,
Se trovi che il tuo prodotto richiede controlli speciali, questo significa che dovrai preparare una presentazione 510(k) alla FDA e ricevere l’autorizzazione prima di andare sul mercato. Successivamente, è necessario registrare il proprio stabilimento ed elencare il prodotto.
Se trovi che il tuo prodotto richiede l’approvazione premarket, questo significa che dovrai seguire il processo FDA PMA per ricevere l’approvazione prima di andare sul mercato.,
Classificazione dei dispositivi medici in Europa

Commissione Europea
I regolamenti per un dispositivo medico nell’Unione Europea (UE) sono stabiliti attraverso le Direttive sui dispositivi medici della Commissione Europea (CE).
La strada verso il mercato in Europa è quella di ottenere una marcatura CE.
Per capire cosa è necessario per ottenere una marcatura CE del dispositivo medico, è necessario prima determinare la classificazione UE del dispositivo medico., Il regolamento dell’Unione Europea sui dispositivi medici (EU MDR) include le informazioni necessarie per determinare la classe del dispositivo.
EU MDR 2017/745 modifica la direttiva 2001/83/CE, il regolamento (CE) n.178/2002 e il regolamento (CE) n. 1223/2009 e abroga le direttive del Consiglio 90/385/CEE e 93/42 / CEE. L’MDR dell’UE diventerà il regolamento obbligatorio per i dispositivi medici a partire da maggio 2020.
È necessario determinare se il dispositivo medico è:
- Non invasivo
- Qualsiasi dispositivo che non penetra nel corpo attraverso un orifizio o la superficie del corpo., Questi dispositivi sono in genere di classe I; tuttavia, si applicano alcune regole ed eccezioni che potrebbero renderli di classe II o superiore.
- Invasivo
- Qualsiasi dispositivo che, in tutto o in parte, penetra all’interno del corpo, attraverso un orifizio del corpo o attraverso la superficie del corpo.
- Attivo
- Qualsiasi dispositivo il cui funzionamento dipende da una fonte di energia diversa da quella generata dal corpo umano a tale scopo, o dalla gravità, e che agisce modificando la densità o convertendo tale energia.,
Per ciascuna delle grandi categorie si applicano alcune regole, descritte nell’allegato VIII del nuovo regolamento sui dispositivi medici. Queste categorie accoppiate con la durata dell’uso rendono la determinazione della classificazione abbastanza semplice.
Ad esempio, un dispositivo in uso continuo per meno di 60 minuti è considerato di durata transitoria, da 60 minuti a 30 giorni è considerato a breve termine e oltre 30 giorni è considerato a lungo termine.,
Con questo in mente, per determinare la classificazione UE del dispositivo, possiamo utilizzare l’esempio di catetere percutaneo utilizzato in precedenza in questa guida per la classificazione FDA.
Diciamo che determino che il mio dispositivo medico si inserisce nella categoria “invasiva”; questo restringe la mia ricerca alle regole 5, 6, 7 e 8.
Posso quindi restringere ulteriormente le regole poiché so che il mio dispositivo medico è a breve termine perché viene utilizzato per un periodo superiore a 24 ore e inferiore a 30 giorni.
Da lì, sono in grado di determinare che la regola 7 è più applicabile alla classificazione del mio dispositivo.,

Fonte: EU MDR 2017/745 Annex VIII 5.3
Poiché il mio catetere percutaneo somministrerà medicinali, posso confermare che il mio dispositivo medico è considerato un dispositivo medico di Classe IIb nel mercato europeo.
DETERMINARE IL TUO PERCORSO VERSO IL MERCATO IN EUROPA
L’Unione Europea ha un sistema di classificazione dei prodotti simile a quello degli Stati Uniti.,:
- Classe I
- Classe IIa
- Classe IIb
- Classe III
In tutti i casi per i dispositivi medici da vendere nell’Unione Europea, la documentazione tecnica è una fase necessaria nel processo di ottenimento della marcatura CE. Tutte le classi di dispositivi medici nell’UE richiedono la collaborazione di un Organismo notificato, ad eccezione di quelle di classe I che possono essere autocertificate.
Le aziende dovranno anche lavorare con un rappresentante autorizzato per occuparsi della registrazione dei prodotti in Europa., Le domande principali dell’articolo 11 sui rappresentanti autorizzati CE possono contribuire a fornire ulteriori chiarimenti su come ottenere la classificazione dei dispositivi medici in Europa.
Classificazione dei dispositivi medici in Canada
![]()
Health Canada
Le normative sui dispositivi medici in Canada sono stabilite dal governo del Canada e regolamentate da Health Canada.
Come negli Stati Uniti e nell’UE, per vendere sul mercato canadese, è necessario innanzitutto determinare la classificazione dei dispositivi medici ai sensi del regolamento canadese.,
Analogamente ai requisiti delineati nell’MDR dell’UE, Health Canada fornisce una guida abbastanza semplice e facile da seguire sul sistema di classificazione basato sul rischio per i dispositivi diagnostici non in vitro da utilizzare per i produttori di dispositivi medici quando vendono in questo mercato.,
Health Canada definisce quattro gruppi di non-dispositivi medico diagnostici in vitro:
- Dispositivi Invasivi (regolamento 1 – 3)
- Non-invasività (Regole 4 – 7)
- Dispositivi Attivi (Regole 8 – 12)
- Particolari regole (Regole di 13 – 16)
Per ciascuna delle categorie, ci sono una serie di regole che si applicano. Queste regole sono ciò che i produttori dovrebbero seguire per determinare la classificazione del rischio del loro dispositivo.
Utilizzerò ancora una volta l’esempio del catetere percutaneo nel caso di commercializzazione di un dispositivo del genere in Canada.,
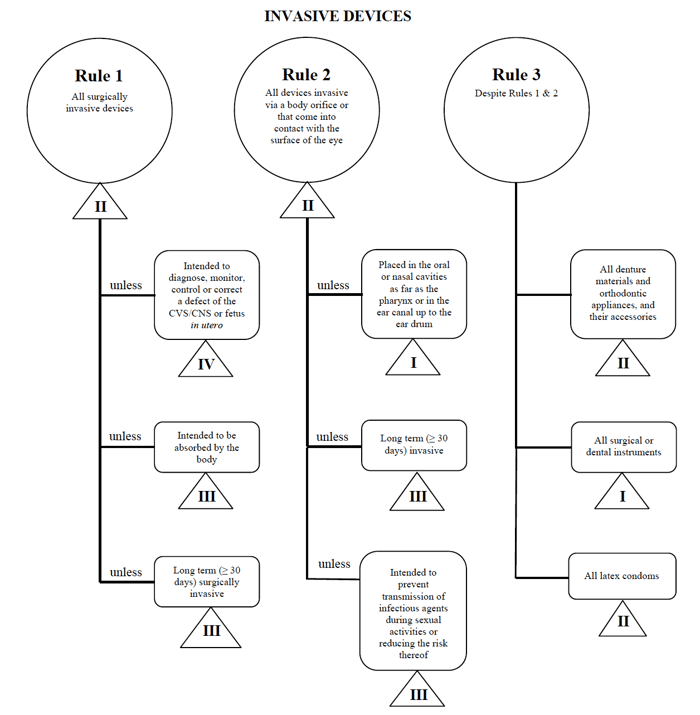
Fonte: Guidance on the Risk based Classification System for Non-In Vitro Diagnostic Devices
I determine my medical device fits into the “invasive” category, restring my search down to Rules 1, 2, and 3.
Dopo aver esaminato le opzioni, stabilisco che si applica la regola 1.
In base all’uso previsto, il mio dispositivo medico è considerato di classe II in Canada.,
DETERMINARE IL PERCORSO DI MERCATO IN CANADA
Ci sono quattro livelli di dispositivo medico classificazioni in Canada:
- Classe I
- Classe II
- Classe III
- Classe IV
Prima di andare al mercato in Canada, è necessario applicare per un dispositivo medico di licenza. I dispositivi medici di classe I non richiedono una licenza. I produttori possono fare riferimento al documento di orientamento Health Canada, che ti guida attraverso questo processo.,
I produttori di dispositivi medici di classe III e Classe IV possono ricevere la loro licenza inviando una domanda di premarket, nei formati ToC o Health Canada, per entrare nel mercato canadese.
È inoltre necessario ottenere la certificazione ISO 13485 con MDSAP.
Update to Health Canada Regulations: MDSAP
A partire da gennaio 2019, tutti i produttori di dispositivi medici che vendono dispositivi medici di classe II e superiori al mercato canadese devono far parte del Medical Device Single Audit Programme (MDSAP).,
Questi produttori devono sottoporsi e superare un audit completo del loro sistema di gestione della qualità (SGQ) attraverso il programma in Canada. Ci sono attualmente 6 regioni in tutto il mondo che partecipano a MDSAP, tra cui Canada, Stati Uniti, Giappone, Brasile e Australia.
Oltre a quei produttori richiesti da Health Canada per partecipare al programma, la partecipazione a MDSAP è facoltativa per i produttori.,
Per ottenere MDSAP di certificazione, i produttori di dispositivi medici devono completare i seguenti tre passaggi:
- Applicazione e recensione
- Off-sito di documentazione di audit
- audit On-site
i produttori di dispositivi di vendita in Canada sarà sottoposto a revisione annuale, con un audit di ricertificazione ogni terzo anno. La conformità con il programma di audit unico dei dispositivi medici si basa sul rispetto delle linee guida della norma ISO 13485 sui sistemi di gestione della qualità dei dispositivi medici.,
Ti consigliamo di formare i tuoi team di prodotto, qualità e normative sui requisiti MDSAP applicabili per semplificare la tua applicazione.
Il nostro strumento di valutazione gap gratuito per MDSAP e ISO 13485 aiuta i produttori di dispositivi a valutare le linee guida sul SGQ dello standard ISO insieme ai requisiti delle organizzazioni di audit Health Canada (AO) e di altre regioni che partecipano al programma.,
Considerazioni finali
Ora hai le informazioni e le risorse necessarie per determinare la classificazione dei dispositivi medici e il percorso di commercializzazione nei tre mercati più grandi del mondo.
Indipendentemente dalla classificazione del dispositivo, è imperativo che si seguono le linee guida normative che si applicano in ogni mercato. Soddisfare la conformità è un aspetto chiave della gestione della qualità che alla fine deciderà il destino del dispositivo e dell’azienda nel suo complesso.,
A Greenlight Guru, apprezziamo l’importanza della gestione della qualità dei dispositivi medici (MDQMS). Il nostro software MDQMS è l’unica soluzione al mondo progettata specificamente per i dispositivi medici. È allineato con le più recenti best practice standard del settore per la gestione dei controlli e dei rischi di progettazione del prodotto, nonché delle attività di controllo delle modifiche e di altri eventi di qualità, fornendo una tracciabilità completa per tutto il ciclo di vita del dispositivo medico.
Stai cercando una soluzione di controllo della progettazione per aiutarti a portare dispositivi medici più sicuri sul mercato più velocemente e con meno rischi?, Clicca qui per fare un rapido tour del software QMS per dispositivi medici di Greenlight Guru →