
o que estou prestes a partilhar convosco é um guia para a classificação regulamentar dos dispositivos médicos.
neste guia, vou fornecer-lhe uma abordagem passo-a-passo para determinar como o seu dispositivo médico será classificado pela FDA DOS EUA, A Comissão Europeia e o Canadá da Saúde. Obter uma compreensão básica da classificação regulamentar de produtos será inestimável para os seus esforços para trazer novos produtos para o mercado.,
classificação regulamentar 101
as regras que se aplicam ao seu dispositivo médico dependem de como o seu produto é classificado pelas agências reguladoras. Cada agência reguladora definiu várias classificações diferentes para dispositivos médicos.
As classificações estão, na maioria das vezes ou regra geral, relacionadas com o risco percebido do tipo de produto.os fabricantes de dispositivos médicos que vendem a nível internacional precisam de se familiarizar com a regulamentação aplicável desses mercados. Isto é mais fácil de dizer do que fazer e pode ser um desafio para a maioria dos fabricantes., Os EUA têm o seu conjunto de regras, enquanto o Canadá adere a outra, e a Europa a outra ainda.Felizmente, Existem muitos paralelos entre os regulamentos e normas internacionais de dispositivos médicos. Este guia foi projetado para mostrar como classificar seu dispositivo em diferentes mercados ao redor do mundo.

por que a classificação regulamentar ainda importa?a classificação do produto determinará o que deve fazer antes de poder vender o seu produto.,a classificação do produto irá ajudá-lo a estabelecer requisitos durante a fase de desenvolvimento do produto, especificamente controlos de concepção.
O seguinte conteúdo não é um guia abrangente para submissões regulamentares, mas deve dar-lhe alguma orientação básica e direção sobre a identificação de como estabelecer caminho para o mercado.vou ficar-me pelo “big 3” que devias saber quando se trata de classificação de dispositivos médicos:, Comida & Administração de Drogas, Centro para Dispositivos & Saúde Radiológica (FDA CDRH)
Dispositivo Médico Regulamentar de Classificação nos EUA

U.S. FOOD & DRUG ADMINISTRATION (FDA)
Nos Estados Unidos, dispositivos médicos, são regulados pela Comida & Administração do Fármaco, ou do FDA., The specific branch within the FDA is the Center for Devices & Radiological Health (CDRH).a missão do CDRH é proteger e promover a saúde pública. Por outras palavras, garantir a segurança dos dispositivos médicos. Nos Estados Unidos, os dispositivos médicos são de classe I, Classe II ou classe III. a classificação de CDRH da FDA é baseada principalmente no risco que o dispositivo médico representa. os dispositivos médicos de classe I são geralmente considerados de baixo risco e os dispositivos médicos de classe III são considerados de maior risco. Os tipos de controlos necessários dependem da classificação do seu produto.,a classificação está directamente relacionada com a utilização pretendida e com as indicações de Utilização. A distinção entre estes termos é um pouco confusa.
- o uso pretendido é o objectivo geral do dispositivo médico ou a sua função (o que você “alega” o dispositivo médico faz).as indicações de Utilização descrevem a doença ou a condição que o dispositivo médico irá diagnosticar, tratar, prevenir, curar ou mitigar, incluindo uma descrição da população-alvo de doentes.
tenha isto em mente., A utilização pretendida e as indicações para a utilização do seu dispositivo médico expressam a razão pela qual teve esta ideia de um novo dispositivo médico.
como encontrar os regulamentos aplicáveis da FDA para o seu dispositivo médico
Uma vez que você define o uso pretendido e as indicações para o uso, Agora você precisa encontrar os possíveis regulamentos e códigos de produto. Rastrear a classificação regulamentar do seu produto através da FDA leva um pouco de tempo e perseverança.,
Sem aborrecê – lo com demasiados detalhes, a FDA estabeleceu várias categorias gerais baseadas na especialidade médica no título 21 da CFR-alimentos e medicamentos: partes 862 a 892.
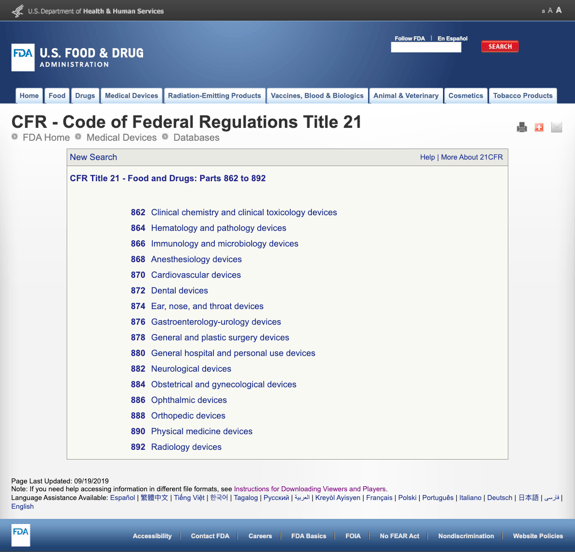
Quando se encontram as categorias possíveis e clicam no número de regulação da FDA, a lista de possibilidades subitamente parece interminável. Aqui está uma visão parcial das opções para a parte 870 dispositivos cardiovasculares:

isto pode ser frustrante e esmagador.,
Quando você encontrar um regulamento que parece ser um ajuste possível, você pode clicar no link e obter mais detalhes para fazer uma determinação.
Por exemplo, se eu acho que o meu dispositivo se encaixa no “870.1250 cateter Percutâneo,” eu clique no link e obter esta informação:

Os detalhes fornecidos me dar alguma idéia se a minha intenção de uso e indicações para uso alinhar com este regulamento específico. Também descobri a classificação do dispositivo FDA.,
neste exemplo, eu aprendo que meu produto é um dispositivo médico de classe II (padrões de desempenho), o que significa que eu vou precisar submeter um 510(k) para a FDA antes de obter a liberação de mercado. Eu compartilho mais sobre os tipos de submissões da FDA mais adiante neste guia.
de seguida-encontre os códigos do produto aplicáveis ao seu dispositivo
encontrando o regulamento aplicável para o seu dispositivo médico e a classificação é a primeira parte. Agora você precisa encontrar os códigos de produto aplicáveis. Aqui está como:
Go to the FDA Product Classification Database and type in the regulation number you found., Se você encontrar mais de uma possibilidade, então você precisará repetir este processo para cada um.
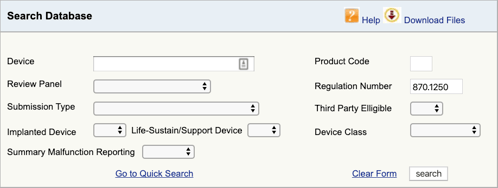
Quando carregar em” search”, irá obter uma lista de possíveis códigos de produtos.
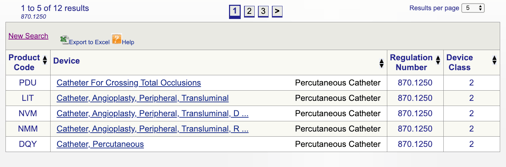
pode então rever cada código individual para determinar a melhor opção para o seu produto clicando em cada código.
determinar o seu caminho para o mercado nos EUA
conhecer a regulamentação aplicável e o código do produto (como descrito acima) é necessário para que você determine a classificação do seu dispositivo médico., Uma vez que você tenha esta informação, você agora será capaz de determinar o “caminho” para obter o seu produto registrado com a FDA.,
FDA define três controlos regulamentares para cada dispositivo médico classe:
- Classe de dispositivos médicos (de baixo a moderado risco): Controles Gerais
- dispositivo médico Classe II (moderado a alto risco): Controles Gerais e Especiais de Controles
- dispositivo médico Classe III (alto risco): Controles Gerais e Premarket Aprovação (PMA)
Deixe-me reduzi-las a este:
Se você encontrar o seu produto é “isenta”, somente controles gerais de aplicar e não formal FDA apresentação é necessária. Você precisa, no entanto, registrar seu estabelecimento com a FDA e, em seguida, listar o produto.,
Se achar que o seu Produto Necessita de controlos especiais, isto significa que terá de preparar uma submissão 510(k) à FDA e receber autorização antes de entrar no mercado. Depois disso, você precisa registrar seu estabelecimento e listar o produto.
Se encontrar que o seu Produto Necessita de aprovação pré-mercado, isto significa que terá de seguir o processo PMA da FDA para receber aprovação antes de entrar no mercado.,
Dispositivo Médico Classificação na Europa

Comissão Europeia
Os regulamentos para um dispositivo médico na União Europeia (UE) são estabelecidas por meio de Directivas relativas aos Dispositivos Médicos pela Comissão Europeia (CE).
O caminho para o mercado na Europa é obter uma marcação CE.para descobrir o que é necessário para obter a marcação CE do seu dispositivo médico, deve primeiro determinar a classificação UE do seu dispositivo médico., O regulamento da União Europeia relativo aos dispositivos médicos (EU MDR) inclui a informação necessária para determinar a sua classe de dispositivo. A Eu MDR 2017/745 altera a Diretiva 2001/83/CE, o Regulamento (CE) n. o 178/2002 e o Regulamento (CE) n. o 1223/2009 e revoga as diretivas 90/385/CEE e 93/42 / CEE do conselho. O MDR da UE passará a ser o regulamento obrigatório para os dispositivos médicos a partir de Maio de 2020.terá de determinar se o seu dispositivo médico é:
- Não Invasivo
- qualquer dispositivo que não penetre o corpo através de um orifício ou da superfície do corpo., Estes dispositivos são tipicamente Classe I; no entanto, certas regras e exceções se aplicam que podem torná-los Classe II ou superior. qualquer dispositivo que, no todo ou em parte, penetre no interior do corpo, quer através de um orifício do corpo, quer através da superfície do corpo.
- Activo
- Qualquer dispositivo cuja operação depende de uma fonte de energia diferente da gerada pelo corpo humano, para essa finalidade, ou pela gravidade, e que atua alterando a densidade ou a conversão de energia.,para cada uma das grandes categorias, existem certas regras aplicáveis, descritas no Anexo VIII do novo regulamento relativo aos dispositivos médicos. Estas categorias, juntamente com a duração de Utilização, tornam a classificação bastante simples. por exemplo, um dispositivo em uso contínuo durante menos de 60 minutos é considerado de duração transitória, 60 minutos a 30 dias é considerado de curto prazo e mais de 30 dias é considerado de longo prazo.,com isso em mente, para determinar a classificação da UE do seu dispositivo, podemos usar o exemplo do cateter percutâneo usado anteriormente neste guia para a classificação da FDA.digamos que determino que o meu dispositivo médico se encaixa na categoria “invasiva”, o que reduz a minha busca às regras 5, 6, 7 e 8. posso então reduzir ainda mais as regras, pois sei que o meu dispositivo médico é de curta duração porque é utilizado por um período superior a 24 horas e inferior a 30 dias.a partir daí, sou capaz de determinar que a regra 7 é mais aplicável à classificação do meu dispositivo.,

Fonte: UE MDR 2017/ 745 Anexo VIII 5.3
Desde a minha percutânea de cateter administrar medicamentos, posso confirmar que a minha médica dispositivo é considerado como uma Classe IIb de dispositivos médicos no mercado Europeu.
determinar o seu caminho para o mercado na Europa
A União Europeia tem um sistema de classificação de produtos semelhante ao dos EUA.,:
- Classe I
- Classe IIa
- Classe IIb
- Classe III
Em todos os casos para os dispositivos médicos para ser vendido na União Europeia, a documentação técnica é um passo necessário no processo de obtenção da Marcação CE. Todas as classes de dispositivos médicos na UE exigem trabalhar com um organismo notificado, excepto as que são da classe I e podem ser auto-certificadas.as empresas também precisam trabalhar com um representante autorizado para cuidar do registro de produtos na Europa., As perguntas mais importantes do artigo 11º sobre os representantes autorizados da CE podem ajudar a esclarecer melhor a forma de obter a Classificação dos dispositivos médicos na Europa.
Classificação dos dispositivos médicos no Canadá

Health Canada
os regulamentos relativos aos dispositivos médicos no Canadá são estabelecidos pelo Governo do Canadá e regulamentados pela Health Canada.como os EUA e a UE, para vender no mercado canadense, você deve primeiro determinar a classificação de dispositivos médicos sob a regulamentação do Canadá.,semelhante aos requisitos descritos na EU MDR, a Health Canada proporciona uma orientação bastante simples e fácil de seguir sobre o sistema de classificação baseado no risco para dispositivos de diagnóstico Não in Vitro que os fabricantes de dispositivos médicos devem utilizar quando vendem neste mercado.,o Canadá define quatro grupos de dispositivos médicos de diagnóstico Não in vitro: dispositivos invasivos (regras 1 – 3) dispositivos não invasivos (regras 4 – 7) dispositivos activos (regras 8 – 12) Regras especiais (regras 13 – 16) Estas regras são as que os fabricantes devem seguir para determinar a classificação de risco do seu dispositivo.
vou usar o exemplo do cateter percutâneo mais uma vez no caso de comercializar tal dispositivo no Canadá.,
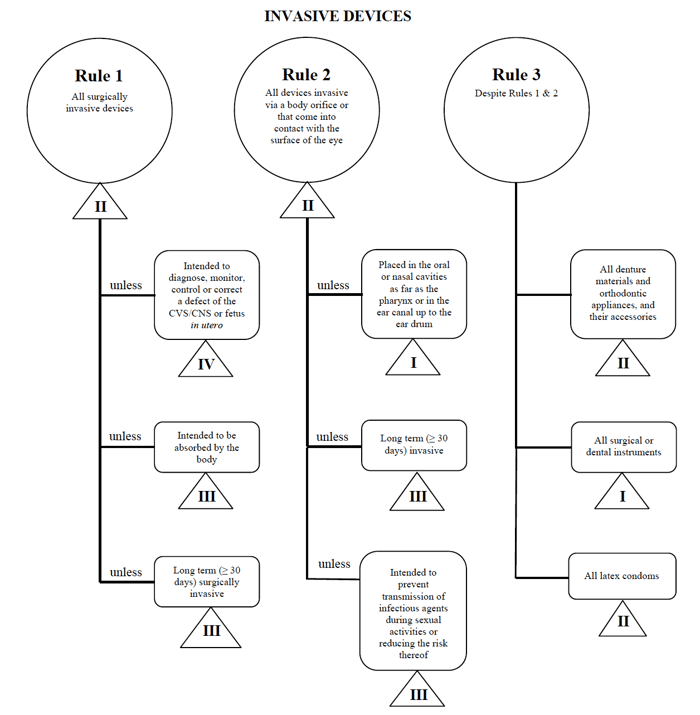
fonte: orientações sobre o sistema de classificação baseado no risco para dispositivos de Diagnóstico não in Vitro
determine que o meu dispositivo médico se encaixa na categoria “invasivo”, restringindo a minha pesquisa às Regras 1, 2 e 3.
Depois de rever as opções, eu determino que a Regra 1 se aplica.com base na utilização pretendida, o meu dispositivo médico é considerado Classe II no Canadá.,
DETERMINAR o SEU CAMINHO PARA o MERCADO NO CANADÁ
Existem quatro níveis de dispositivo médico classificações no Canadá:
- Classe I
- categoria II
- Classe III
- Classe IV
Antes de ir ao mercado em Canadá, você deve primeiro aplicar para um dispositivo médico de licença. Os dispositivos médicos de classe I não necessitam de licença. Os fabricantes podem consultar o documento de orientação Health Canada, que o acompanha neste processo.,
fabricantes de dispositivos médicos de classe III e classe IV podem receber a sua licença através da apresentação de uma aplicação pré-mercado, quer nos formatos ToC ou Health Canada, para entrar no mercado canadense.também terá de obter a certificação ISO 13485 com o MDSAP.
Update to Health Canada Regulations: MDSAP
a partir de janeiro de 2019, todos os fabricantes de dispositivos médicos que vendem dispositivos médicos de classe II e superiores ao mercado canadiano precisam fazer parte do Programa de auditoria única de dispositivos médicos (MDSAP)., estes fabricantes devem submeter-se a uma auditoria completa do seu sistema de gestão da qualidade (QMS) através do programa no Canadá. Existem atualmente 6 regiões em todo o mundo que participam do MDSAP, incluindo Canadá, EUA, Japão, Brasil e Austrália.
além dos fabricantes exigidos pela Health Canada para participar do programa, a participação no MDSAP é opcional para os fabricantes.,para obter a certificação MDSAP, os fabricantes de dispositivos médicos devem completar as três etapas seguintes: aplicação e revisão da auditoria de documentação fora do local auditoria de documentação no local fabricantes de dispositivos que vendem no Canadá serão sujeitos a revisões anuais, com uma auditoria de recertificação de três em três anos. A conformidade com o programa de auditoria única de dispositivos médicos baseia-se no cumprimento das orientações da norma ISO 13485 sobre sistemas de gestão da qualidade para dispositivos médicos.,
recomendamos a formação de suas equipes de produto e qualidade e regulamentação nos requisitos MDSAP aplicáveis para agilizar sua aplicação.a nossa ferramenta de Avaliação livre de lacunas para o MDSAP e ISO 13485 ajuda os fabricantes de dispositivos a avaliar as diretrizes do QMS a partir da norma ISO, juntamente com os requisitos das organizações de auditoria da Health Canada (AO) e de outras regiões que participam no programa.,
Pensamentos finais
agora tem a informação e os recursos de que necessita para determinar a sua classificação e o caminho para o mercado nos três maiores mercados do mundo.
independentemente da classificação de seu dispositivo, é imperativo que você siga as diretrizes regulatórias que se aplicam em cada mercado. Cumprir a conformidade é um aspecto fundamental da Gestão da qualidade que acabará por decidir o destino do seu dispositivo e empresa como um todo.,
no Greenlight Guru, valorizamos a importância da Gestão da qualidade dos dispositivos médicos (MDQMS). Nosso software MDQMS é a única solução no mundo projetado especificamente para dispositivos médicos. Está alinhado com as mais recentes práticas padrão da indústria para a gestão de controles de design de produtos e riscos, bem como as atividades de controle de mudança e outros eventos de qualidade, proporcionando rastreabilidade total ao longo do ciclo de vida de seu dispositivo médico.
à procura de uma solução de controlo de projecto para o ajudar a trazer dispositivos médicos mais seguros para o mercado mais rápido com menos risco?, Clique aqui para fazer uma visita rápida ao Software QMS do dispositivo médico do Greenlight Guru →