
lo que voy a compartir con usted es una guía para la clasificación reguladora de dispositivos médicos.
en esta guía, le proporcionaré un enfoque paso a paso para determinar cómo su dispositivo médico será clasificado por la FDA de los Estados Unidos, la Comisión Europea y Health Canada. Obtener una comprensión básica de la clasificación regulatoria de productos será invaluable para sus esfuerzos por llevar nuevos productos al mercado.,
Clasificación reglamentaria 101
las reglas que se aplican a su dispositivo médico dependen de cómo las agencias reguladoras clasifiquen su producto. Cada agencia reguladora ha definido varias clasificaciones diferentes para dispositivos médicos.
las clasificaciones están, en su mayor parte o por regla general, relacionadas con el riesgo percibido del tipo de producto.
los fabricantes de dispositivos médicos que venden internacionalmente deben familiarizarse con las regulaciones aplicables de esos mercados. Esto es más fácil decirlo que hacerlo y puede ser un desafío para la mayoría de los fabricantes., Los EE.UU. tienen su conjunto de reglas, mientras que Canadá se adhiere a otra, y Europa a otra todavía.
afortunadamente, hay muchos paralelismos entre las regulaciones y estándares internacionales de dispositivos médicos. Esta guía está diseñada para mostrarle cómo clasificar su dispositivo en diferentes mercados de todo el mundo.

¿por Qué Reglamentaria Clasificación Importa Siquiera?
saber cómo se clasifica su dispositivo médico es importante por las siguientes razones:
- La clasificación del producto determinará lo que tiene que hacer antes de poder vender su producto.,
- La clasificación de productos le ayudará a establecer los requisitos durante la fase de desarrollo del producto, específicamente los controles de diseño.
- La clasificación de productos es un componente importante para determinar cuánto costará llevar su dispositivo al mercado y darle una idea de cuánto tiempo tomará.
debido a esto, voy a proporcionarte un poco de orientación para comprender mejor qué hacer y cómo hacerlo.,
Nota, la información que estoy a punto de proporcionar está destinada a ayudarlo a educarlo sobre la clasificación reguladora de dispositivos médicos y lo que se requiere para su dispositivo médico.
el siguiente contenido no es una guía completa para presentaciones regulatorias, sin embargo, debe darle alguna orientación básica y dirección sobre la identificación de cómo establecer el camino al mercado.
Me quedo con el «big 3» que debe saber cuando se trata de la clasificación de dispositivos médicos:
- U. S., Food & Drug Administration, Center for Devices & Radiological Health (FDA CDRH)
- European Commission
- Health Canada
Medical Device Regulatory Classification in the U. S.

U. S. Food & Drug Administration (FDA)
en los Estados Unidos, los dispositivos médicos están regulados por la Food & Drug Administration, o FDA., La rama específica dentro de la FDA es el Centro para dispositivos & Salud Radiológica (CDRH).
La misión de CDRH es proteger y promover la salud pública. En otras palabras, asegúrese de que los dispositivos médicos sean seguros. En los Estados Unidos, los dispositivos médicos son de clase I, clase II o clase III. la clasificación CDRH de la FDA se basa principalmente en el riesgo que plantea el dispositivo médico.
los dispositivos médicos de clase I generalmente se consideran de bajo riesgo y los dispositivos médicos de clase III se consideran de mayor riesgo. Los tipos de controles requeridos dependen de la clasificación de su producto.,
La clasificación está directamente relacionada con el uso previsto y las indicaciones de uso. La distinción entre estos términos es un poco confusa.
- El uso previsto es el propósito general del dispositivo médico o su función (lo que usted «afirma» que hace el dispositivo médico).
- Las indicaciones de uso describen la enfermedad o condición que el dispositivo médico diagnosticará, tratará, prevendrá, curará o mitigará, incluyendo una descripción de la población de pacientes objetivo.
tenga esto en cuenta., El uso previsto y las indicaciones para el uso de su dispositivo médico expresan la razón por la que tuvo esta idea para un nuevo dispositivo médico.
cómo encontrar las regulaciones aplicables de la FDA para su dispositivo médico
Una vez que defina el uso previsto y las indicaciones de uso, ahora necesita encontrar las posibles regulaciones y códigos de producto. El seguimiento de la clasificación reglamentaria de su producto a través de la FDA requiere un poco de tiempo y perseverancia.,
sin aburrirle con demasiados detalles, la FDA ha establecido varias categorías generales basadas en la especialidad médica en el título 21 del CFR – Alimentos y medicamentos: partes 862 a 892.
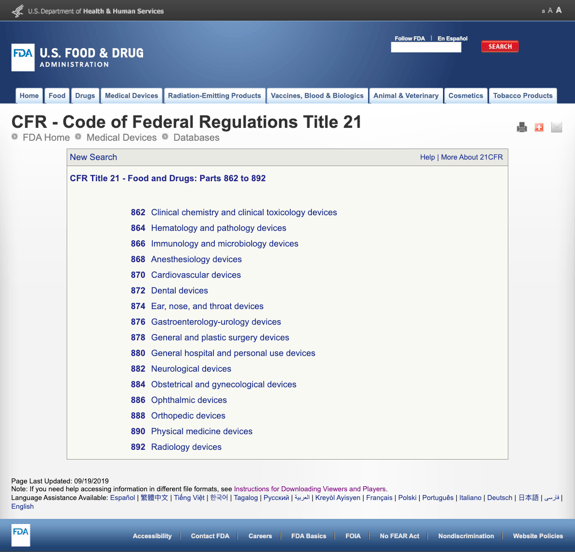
Cuando encuentre las posibles categorías y haga clic en el número de regulación de la FDA, la lista de posibilidades de repente parece interminable. Aquí hay una vista parcial de las opciones para los dispositivos cardiovasculares de la parte 870:

esto puede ser frustrante y abrumador.,
Cuando encuentre una regulación que parezca ser un posible ajuste, puede hacer clic en el enlace y obtener más detalles para hacer una determinación.
por ejemplo, si creo que mi dispositivo encaja en «catéter percutáneo 870.1250», hago clic en el enlace y obtengo esta información:

Los detalles proporcionados me dan alguna idea si mi uso previsto y las indicaciones de uso se alinean con esta regulación específica. También descubro la clasificación del dispositivo de la FDA.,
en este ejemplo, me entero de que mi producto es un dispositivo médico de clase II (estándares de rendimiento), lo que significa que tendré que presentar un 510(k) a la FDA antes de obtener la autorización del mercado. Comparto más información sobre los tipos de presentaciones de la FDA más adelante en esta guía.
a continuación, Encuentre los códigos de Producto aplicables a su dispositivo
encontrar la regulación aplicable para su dispositivo médico y la clasificación es la primera parte. Ahora necesita encontrar los códigos de producto aplicables. Así es como:
vaya a la base de datos de clasificación de productos de la FDA y escriba el número de regulación que encontró., Si encuentra más de una posibilidad, entonces tendrá que repetir este proceso para cada uno.
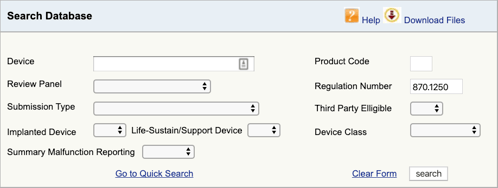
Cuando usted haga clic en «buscar», aparecerá una lista de posibles códigos de producto.
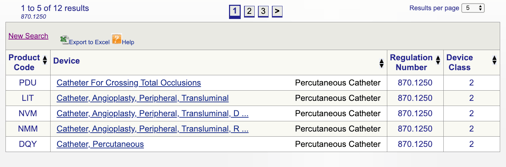
a continuación, puede revisar cada código individual para determinar la mejor opción para su producto haciendo clic en cada código.
determinar su camino al mercado en los EE. UU.
conocer la regulación aplicable y el código de producto (como se describe anteriormente) es necesario para que pueda determinar la clasificación de su dispositivo médico., Una vez que tenga esta información, ahora podrá determinar la «ruta» para registrar su producto con la FDA.,
la FDA define tres controles reglamentarios para cada clase de dispositivo médico:
- dispositivo médico de clase I (riesgo bajo a moderado): controles generales
- dispositivo médico de clase II (riesgo moderado a alto): controles generales y controles especiales
- dispositivo médico de clase III (riesgo alto): controles generales y aprobación previa a la comercialización (PMA)
«entonces solo se aplican controles generales y no se requiere presentación formal de la FDA. Usted, sin embargo, tiene que registrar su establecimiento con la FDA y luego enumerar el producto.,
Si encuentra que su producto requiere controles especiales, esto significa que tendrá que preparar una presentación 510 (k) a la FDA y recibir autorización antes de salir al mercado. Después de eso, es necesario registrar su establecimiento y la lista del producto.
Si encuentra que su producto requiere aprobación previa a la comercialización, esto significa que tendrá que seguir el proceso PMA de la FDA para recibir la aprobación antes de salir al mercado.,
clasificación de Dispositivos Médicos en Europa

Comisión Europea
las regulaciones para un dispositivo médico en la Unión Europea (UE) se establecen a través de las directivas de Dispositivos Médicos de la Comisión Europea (CE).
el camino hacia el mercado en Europa es obtener un marcado CE.
para averiguar qué se requiere para obtener el marcado CE de su dispositivo médico, primero debe determinar la clasificación de la UE de su dispositivo médico., El Reglamento de dispositivos médicos de la Unión Europea (EU MDR) incluye la información necesaria para determinar su clase de dispositivo.
EU MDR 2017/745 modifica la Directiva 2001/83/CE, el Reglamento (CE) No 178/2002 y el Reglamento (CE) no 1223/2009 y deroga las directivas 90/385/CEE y 93/42 / CEE del Consejo. La MDR de la UE se convertirá en la regulación obligatoria para dispositivos médicos a partir de mayo de 2020.
deberá determinar si su dispositivo médico es:
- No Invasivo
- cualquier dispositivo que no penetre en el cuerpo a través de un orificio o la superficie del cuerpo., Estos dispositivos son típicamente Clase I; sin embargo, se aplican ciertas reglas y excepciones que podrían convertirlos en clase II o superior.
- invasivo
- cualquier dispositivo que, total o parcialmente, penetre en el interior del cuerpo, ya sea a través de un orificio corporal o a través de la superficie del cuerpo.
- activo
- cualquier dispositivo cuyo funcionamiento dependa de una fuente de energía distinta de la generada por el cuerpo humano para ese fin, o por gravedad, y que actúe cambiando la densidad de esa energía o convirtiéndola.,
para cada una de las categorías generales, hay ciertas reglas que se aplican, descritas en el Anexo VIII del nuevo Reglamento de dispositivos médicos. Estas categorías, junto con la duración del uso, hacen que la determinación de la clasificación sea bastante sencilla.
por ejemplo, un dispositivo en uso continuo durante menos de 60 minutos se considera una duración transitoria, de 60 minutos a 30 días se considera a corto plazo, y más de 30 días se considera a largo plazo.,
teniendo esto en cuenta, para determinar la clasificación de su dispositivo en la UE, podemos utilizar el ejemplo de catéter percutáneo utilizado anteriormente en esta guía para la clasificación de la FDA.
digamos que determino que mi dispositivo médico encaja en la categoría «invasiva»; esto reduce mi búsqueda a las reglas 5, 6, 7 y 8.
entonces puedo reducir aún más las reglas ya que sé que mi dispositivo médico es a corto plazo porque se usa por un período mayor de 24 horas y menos de 30 días.
a partir de ahí, puedo determinar que la Regla 7 es más aplicable a la clasificación de mi dispositivo.,

fuente: EU MDR 2017/745 Anexo VIII 5.3
dado que mi catéter percutáneo administrará medicamentos, puedo confirmar que mi dispositivo médico se considera un dispositivo médico de clase IIb en el mercado europeo.
determinar su camino hacia el mercado en Europa
La Unión Europea tiene un sistema de clasificación de productos similar al DE LOS ESTADOS UNIDOS.,:
- Class I
- Class IIa
- Class IIb
- Class III
en todos los casos para que los dispositivos médicos se vendan en la Unión Europea, la documentación técnica es un paso requerido en el proceso de obtención del Marcado CE. Todas las clases de dispositivos médicos en la UE requieren trabajar con un organismo notificado, excepto las que son de clase I y pueden autocertificarse.
Las empresas también tendrán que trabajar con un Representante Autorizado para hacerse cargo del registro de productos en Europa., Las principales preguntas del artículo 11 sobre los representantes autorizados de la CE pueden ayudar a proporcionar más aclaraciones sobre cómo lograr la clasificación de dispositivos médicos en Europa.
clasificación de Dispositivos Médicos en Canadá
![]()
Health Canada
Las regulaciones de dispositivos médicos en Canadá están establecidas por el Gobierno de Canadá y reguladas por Health Canada.
al igual que los EE.UU. y la UE, para vender en el mercado canadiense, primero debe determinar la clasificación de dispositivos médicos según la regulación de Canadá.,
Similar a los requisitos descritos en el MDR de la UE, Health Canada proporciona una guía bastante sencilla y fácil de seguir sobre el sistema de clasificación basado en el riesgo para dispositivos de diagnóstico no in Vitro para que los fabricantes de dispositivos médicos los utilicen al vender en este mercado.,
Health Canada define cuatro grupos de dispositivos médicos de diagnóstico no in vitro:
- dispositivos invasivos (reglas 1-3)
- dispositivos no invasivos (reglas 4-7)
- dispositivos activos (reglas 8 – 12)
- reglas especiales (reglas 13 – 16)
para cada una de las categorías generales, hay un conjunto de reglas que se aplican. Estas reglas son las que los fabricantes deben seguir para determinar la clasificación de riesgo de su dispositivo.
voy a utilizar el ejemplo de catéter percutáneo una vez más en el caso de la comercialización de un dispositivo de este tipo en Canadá.,
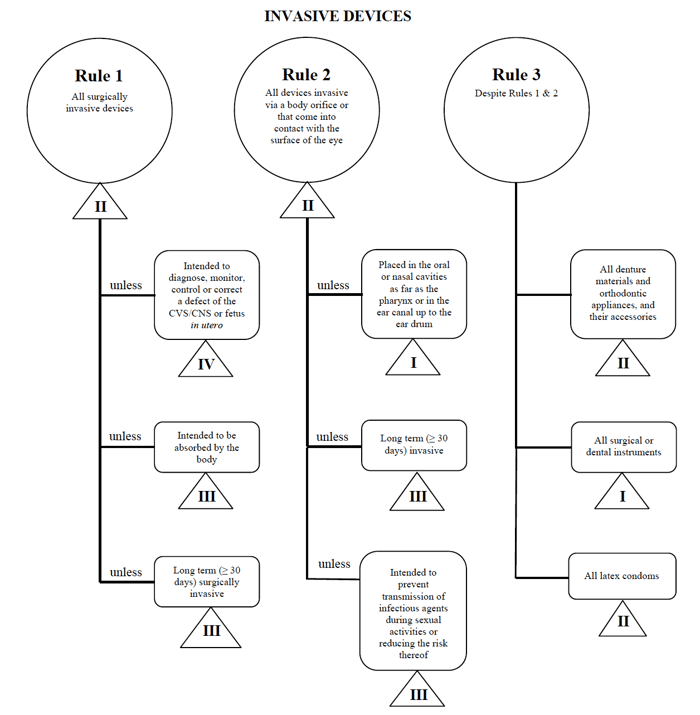
fuente: Guidance on the Risk based Classification System for Non-in Vitro Diagnostic Devices
determino que mi dispositivo médico encaja en la categoría «invasiva», reduciendo mi búsqueda a las reglas 1, 2 y 3.
después de revisar las opciones, determino que se aplica la Regla 1.
según mi uso previsto, Mi dispositivo médico se considera Clase II en Canadá.,
determinar su camino hacia el mercado en Canadá
hay cuatro niveles de clasificaciones de dispositivos médicos en Canadá:
- Clase I
- Clase II
- Clase III
- Clase IV
antes de salir al mercado en Canadá, primero debe solicitar una licencia de dispositivo médico. Los dispositivos médicos de clase I no requieren una licencia. Los fabricantes pueden consultar el documento de orientación de Health Canada, que lo guía a través de este proceso.,
Los fabricantes de dispositivos médicos de clase III y clase IV pueden recibir su licencia enviando una solicitud previa al mercado, ya sea en los formatos ToC O Health Canada, para ingresar al mercado canadiense.
también necesitará obtener la certificación ISO 13485 con MDSAP.
actualización de las regulaciones de Health Canada: MDSAP
a partir de enero de 2019, todos los fabricantes de dispositivos médicos que vendan dispositivos médicos de clase II y superiores al mercado canadiense deben formar parte del Programa de auditoría única de Dispositivos Médicos (MDSAP).,
estos fabricantes deben someterse y pasar una auditoría completa de su sistema de gestión de la calidad (SGC) a través del programa en Canadá. Actualmente hay 6 regiones alrededor del mundo que participan en MDSAP, incluyendo Canadá, Estados Unidos, Japón, Brasil y Australia.
aparte de los fabricantes que Health Canada requiere para participar en el programa, la participación en MDSAP es opcional para los fabricantes.,
para obtener la certificación MDSAP, los fabricantes de dispositivos médicos deben completar los siguientes tres pasos:
- aplicación y revisión
- auditoría de documentación externa
- auditoría in situ
los fabricantes de dispositivos que vendan en Canadá estarán sujetos a revisiones anuales, con una auditoría de recertificación cada tercer año. El cumplimiento del Programa de auditoría única de dispositivos médicos se basa en el cumplimiento de las directrices de la norma ISO 13485 sobre sistemas de gestión de calidad para dispositivos médicos.,
recomendamos capacitar a sus equipos de productos y de calidad y reguladores en los requisitos MDSAP aplicables para agilizar su aplicación.
nuestra herramienta gratuita de evaluación de brechas para MDSAP e ISO 13485 ayuda a los fabricantes de dispositivos a evaluar las directrices de QMS de la norma ISO junto con los requisitos de las organizaciones de auditoría de Health Canada (ao) y otras regiones que participan en el programa.,
Pensamientos finales
ahora tiene la información y los recursos que necesita para determinar su clasificación de dispositivos médicos y su camino al mercado en los tres mercados más grandes del mundo.
independientemente de la clasificación de tu dispositivo, es imperativo que sigas las pautas regulatorias que se aplican en cada mercado. Cumplir con el cumplimiento es un aspecto clave de la gestión de calidad que, en última instancia, decidirá el destino de su dispositivo y de la empresa en su conjunto.,
en Greenlight Guru, valoramos la importancia de la gestión de calidad de dispositivos médicos (MDQMS). Nuestro software MDQMS es la única solución en el mundo diseñada específicamente para dispositivos médicos. Está alineado con las mejores prácticas estándar más recientes de la industria para administrar los controles de diseño de productos y los riesgos, así como las actividades de control de cambios y otros eventos de calidad, proporcionando una trazabilidad completa a lo largo del ciclo de vida de su dispositivo médico.
¿está buscando una solución de control de diseño que lo ayude a comercializar dispositivos médicos más seguros más rápido y con menos riesgo?, Haga clic aquí para hacer un recorrido rápido por el software QMS de Dispositivos Médicos de Greenlight Guru →