
Co se s vámi Chystám sdílet, je průvodce regulatorní klasifikací zdravotnických prostředků.
v této příručce vám poskytnu krok za krokem přístup k určení, jak bude váš zdravotnický prostředek klasifikován americkou FDA, Evropskou komisí a Health Canada. Získání základní znalosti o klasifikaci regulačních produktů bude neocenitelné pro vaše úsilí o uvedení nových produktů na trh.,
regulační klasifikace 101
pravidla, která se vztahují na váš zdravotnický prostředek, závisí na tom, jak je váš produkt klasifikován regulačními agenturami. Každá regulační agentura definovala několik různých klasifikací zdravotnických prostředků.
klasifikace se většinou nebo obecně vztahují k vnímanému riziku typu výrobku.
Výrobci zdravotnických prostředků prodávajících na mezinárodní úrovni se musí seznámit s platnými předpisy těchto trhů. To se snadněji řekne, než udělá, a může být výzvou pro většinu výrobců., USA mají svá pravidla, zatímco Kanada dodržuje další a Evropa ještě další.
Naštěstí Existuje mnoho paralel mezi mezinárodními předpisy a standardy zdravotnických prostředků. Tato příručka je navržena tak, aby vám ukázala, jak klasifikovat Zařízení na různých trzích po celém světě.

proč na regulační klasifikaci vůbec záleží?
vědět, jak je váš zdravotnický prostředek klasifikován, záleží na následujících důvodech:
- klasifikace produktů určí, co musíte udělat, než budete moci produkt prodat.,
- klasifikace produktů vám pomůže stanovit požadavky během fáze vývoje produktu, konkrétně návrhové kontroly.
- klasifikace produktů je důležitou součástí při určování toho, kolik bude stát uvedení vašeho zařízení na trh a dá vám představu o tom, jak dlouho to bude trvat.
kvůli tomu Vám poskytnu trochu pokynů, abyste lépe porozuměli tomu, co dělat a jak to udělat.,
Poznámka: informace, které se chystám poskytnout, jsou určeny k tomu, aby vám pomohly vzdělávat regulační klasifikaci zdravotnických prostředků a to, co je požadováno pro váš zdravotnický prostředek.
následující obsah je komplexní průvodce regulační podání, přesto by se vám některé základní pokyny a směr na zjišťování, jak vytvořit cestu k trhu.
budu se držet „big 3“, který byste měli vědět, pokud jde o klasifikaci zdravotnických prostředků:
- USA., Jídlo & Drug Administration, Centrum pro Zařízení & Radiologické Zdraví (FDA CDRH)
- Evropská Komise
- Zdraví Kanada
Zdravotnické Zařízení, Regulační Klasifikace v USA

US FOOD & DRUG ADMINISTRATION (FDA)
Ve Spojených Státech, zdravotnické prostředky jsou regulovány Food & Drug Administration, nebo FDA., Specifické odvětví v rámci FDA je Centrum pro zařízení & radiologické zdraví (CDRH).
posláním CDRH je chránit a podporovat veřejné zdraví. Jinými slovy, zajistěte, aby zdravotnické prostředky byly bezpečné. V USA jsou zdravotnické prostředky buď třídy I, třídy II nebo třídy III. klasifikace FDA CDRH je založena především na riziku, které představuje zdravotnický prostředek.
zdravotnické prostředky třídy I jsou obecně považovány za nízké riziko a zdravotnické prostředky třídy III jsou považovány za nejvyšší riziko. Typy požadovaných kontrol závisí na klasifikaci vašeho produktu.,klasifikace
přímo souvisí s zamýšleným použitím a indikací pro použití. Rozdíl mezi těmito pojmy je trochu matoucí.
- zamýšlené použití je obecným účelem zdravotnického prostředku nebo jeho funkce(to, co „tvrdí“ zdravotnický prostředek).
- Indikace pro Použití popsat onemocnění nebo stav, lékařské zařízení se diagnostice, léčbě, prevenci, vyléčení nebo zmírnění, včetně popisu cílové populaci pacientů.
Mějte to na paměti., Zamýšlené použití a indikace pro použití vašeho zdravotnického prostředku vyjadřují důvod, proč jste měli tento nápad na nový zdravotnický prostředek.
jak najít platné předpisy FDA pro váš zdravotnický prostředek
jakmile definujete zamýšlené použití a indikace pro použití, musíte nyní najít možné předpisy a kódy produktů. Sledování regulační klasifikace pro váš produkt prostřednictvím FDA trvá trochu času a vytrvalosti.,
aniž by vás nudil příliš mnoha detaily, FDA vytvořila několik obecných kategorií založených na lékařské specialitě v hlavě CFR 21 – potraviny a drogy: části 862 až 892.
Když najdete možné kategorie a kliknete na číslo regulace FDA, seznam možností se náhle zdá nekonečný. Zde je částečný pohled na možnosti pro Část 870 Kardiovaskulární zařízení:

To může být frustrující a ohromující.,
Když zjistíte, nařízení, které se zdá být možné přizpůsobit, můžete kliknout na odkaz a získat více informací, aby se rozhodnutí.
například, pokud si myslím, že můj přístroj se vejde do „870.1250 Perkutánní katétr,“ jsem kliknout na odkaz a získat tyto informace:

podrobnosti za předpokladu, dej mi nějaký nápad, jestli můj určené použití a indikace pro použití sladit s touto konkrétní nařízení. Také objevuji klasifikaci zařízení FDA.,
v tomto příkladu se dozvídám, že můj produkt je zdravotnický prostředek třídy II (výkonnostní standardy), což znamená, že budu muset předložit FDA 510(k) před získáním povolení na trhu. Sdílím více o typech podání FDA dále v této příručce.
Next-Najděte Kódy produktů platné pro vaše zařízení
Najděte příslušné nařízení pro zdravotnické zařízení a klasifikace je první část. Nyní musíte najít příslušné kódy produktů. Zde je návod:
přejděte do databáze klasifikace produktů FDA a zadejte číslo předpisu, které jste našli., Pokud zjistíte více než jednu možnost, budete muset tento proces opakovat pro každou z nich.
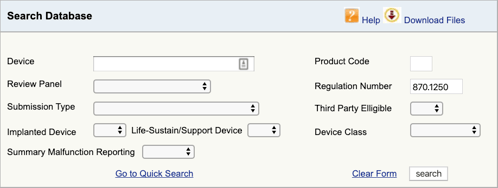
po kliknutí na tlačítko“ Hledat “ získáte seznam možných kódů produktů.
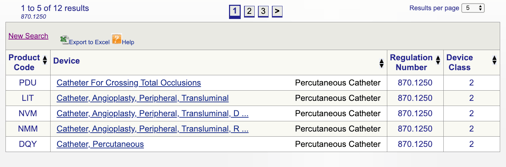
poté můžete zkontrolovat každý jednotlivý kód a určit nejlepší možnost pro váš produkt kliknutím na každý kód.
určení vaší cesty na trh v USA
Znalost platného nařízení a kódu produktu (jak je popsáno výše) je nezbytná pro určení klasifikace vašeho zdravotnického prostředku., Jakmile budete mít tyto informace, budete nyní moci určit „cestu“, aby se váš produkt registrován u FDA.,
FDA definuje tři regulační kontroly pro každý zdravotnický prostředek třídy:
- Třída zdravotnického přístroje (nízké až střední riziko): Obecné Ovládací prvky
- Třídy II zdravotnických zařízení (střední až vysoké riziko): Obecné Ovládací prvky a Speciální Ovládací prvky
- III. Třída zdravotnické zařízení (vysoké riziko): Obecné Ovládací prvky a Premarket Schválení (PMA)
Dovolte mi, abych šel do tohoto:
Pokud zjistíte, že váš produkt je „osvobozen“, pak pouze obecné ovládací prvky použít a žádné formální FDA předložení je požadováno. Musíte však zaregistrovat své zařízení u FDA a poté uvést produkt.,
Pokud zjistíte, že váš produkt vyžaduje speciální ovládací prvky, znamená to, že budete muset připravit 510(k) podání FDA a získat povolení před odchodem na trh. Poté musíte zaregistrovat své zařízení a uvést produkt.
Pokud zjistíte, že váš produkt vyžaduje schválení premarket, to znamená, že budete muset sledovat proces FDA PMA získat schválení před odchodem na trh.,
Zdravotnické Zařízení Klasifikace v Evropě

Evropská Komise
předpisy pro lékařská zařízení v Evropské Unii (EU) jsou stanoveny prostřednictvím Zdravotnických Zařízení Směrnice Evropské Komise (EK).
cesta na trh v Evropě je získat označení CE.
Chcete-li zjistit, co je zapotřebí k získání označení CE vašeho zdravotnického prostředku, musíte nejprve určit klasifikaci vašeho zdravotnického prostředku v EU., Nařízení Evropské unie o zdravotnických prostředcích (EU MDR) obsahuje potřebné informace k určení třídy zařízení.
EU MDR 2017/745 mění směrnici 2001/83 / ES, nařízení (ES) č. 178/2002 a nařízení (ES) č. 1223/2009 a zrušuje směrnice Rady 90/385/EHS a 93/42/EHS. EU MDR se od května 2020 stane povinnou regulací zdravotnických prostředků.
budete muset určit, zda je váš zdravotnický prostředek:
- neinvazivní
- jakékoli zařízení, které neproniká tělem otvorem nebo povrchem těla., Tato zařízení jsou obvykle třída I; platí však určitá pravidla a výjimky, které by je mohly učinit třídou II nebo vyšší.
- Invazivní
- Jakékoliv zařízení, které zcela nebo zčásti proniká do těla, buď tělním otvorem nebo povrchem těla.
- Aktivní
- Jakékoliv zařízení, jehož provoz závisí na zdroji energie, jiných než, že generovaná lidským tělem pro tento účel, nebo gravitací, a který působí prostřednictvím změny hustoty nebo přeměny této energie.,
pro každou ze širokých kategorií platí určitá pravidla uvedená v příloze VIII nového předpisu o zdravotnických prostředcích. Tyto kategorie spolu s dobou používání činí určování klasifikace poměrně přímočaré.
například, zařízení je v nepřetržitém používání za 60 minut je považován za přechodné trvání, 60 minut až 30 dní, je považován za krátkodobý, a více než 30 dní, je považován za dlouhodobý.,
s ohledem na to, abychom určili klasifikaci vašeho zařízení v EU, můžeme použít příklad perkutánního katétru použitý dříve v této příručce pro klasifikaci FDA.
řekněme, že určím, že můj zdravotnický prostředek zapadá do kategorie“ invazivní“; to zužuje mé vyhledávání na pravidla 5, 6, 7 a 8.
poté mohu pravidla dále zúžit, protože vím, že můj zdravotnický prostředek je krátkodobý, protože se používá po dobu delší než 24 hodin a kratší než 30 dní.
odtud jsem schopen určit, že pravidlo 7 je nejvíce použitelné pro klasifikaci mého zařízení.,

Zdroj: EU MDR 2017/ 745 Příloha VIII 5.3
Protože moje perkutánní katétr bude podávat léky, mohu potvrdit, že můj zdravotní zařízení je považováno za Třídy IIb zdravotnické zařízení na Evropském trhu.
určení vaší cesty na trh v Evropě
Evropská unie má podobný systém klasifikace produktů jako USA.,:
- Třída
- Třída
- Třídy IIb
- III. Třídy
Ve všech případech pro lékařská zařízení, které mají být prodávány v Evropské Unii, technická dokumentace je povinný krok v procesu získání Označení CE. Všechny třídy zdravotnických prostředků v EU vyžadují práci s oznámeným subjektem, s výjimkou těch, které jsou třídy I a mohou být certifikovány Samostatně.
společnosti budou také muset spolupracovat s oprávněným zástupcem, aby se postaraly o registraci produktů v Evropě., Článek 11 hlavní otázky týkající se oprávněných zástupců ES mohou pomoci poskytnout další vysvětlení, jak dosáhnout klasifikace zdravotnických prostředků v Evropě.
Zdravotnické Zařízení Klasifikace v Kanadě
![]()
Zdraví Kanada
Na zdravotnické prostředky nařízení v Kanadě jsou zřízeným Vládou Kanady a regulována Health Canada.
jako USA a EU, Chcete-li prodat na kanadském trhu, musíte nejprve určit klasifikaci zdravotnických prostředků podle kanadského nařízení.,
Podobné požadavky uvedené v EU MDR, Health Canada poskytuje poměrně jednoduché a snadno sledovat Pokyny na Rizika na základě Systému Klasifikace pro Non-Diagnostické Prostředky In Vitro pro lékařské výrobci zařízení používat při prodeji na tomto trhu.,
Zdraví Kanady definuje čtyři skupiny non-diagnostické zdravotnické prostředky in vitro:
- Invazivní Prostředky (Pravidla 1 – 3)
- Non-Invazivní Prostředky (Pravidla 4 – 7)
- Aktivní Zařízení (Pravidla 8 – 12)
- Speciální Pravidla (13 – 16)
Pro každou z hlavních kategorií, existuje sada pravidel, která platí. Tato pravidla jsou to, co by výrobci měli dodržovat, aby určili klasifikaci rizik svého zařízení.
v případě uvedení takového zařízení na trh v Kanadě použiji příklad perkutánního katétru.,
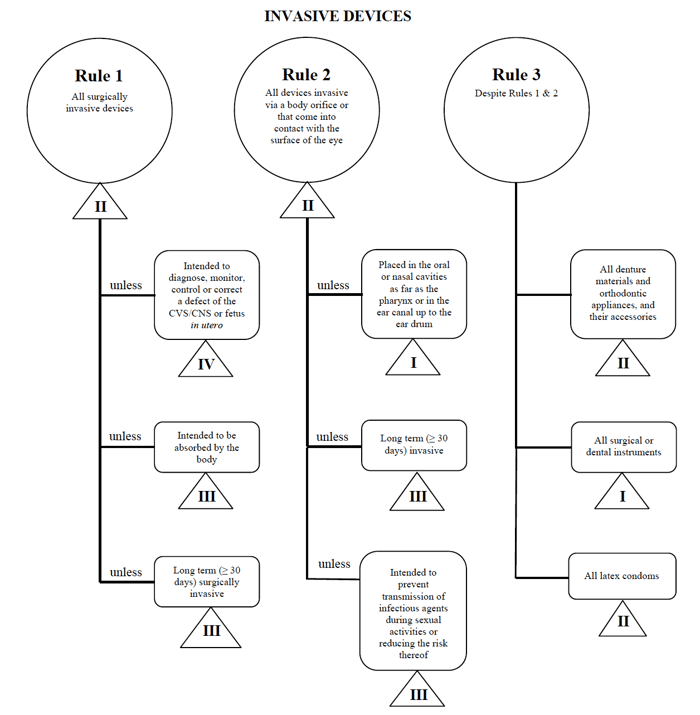
Zdroj: Návod na Rizika na základě Systému Klasifikace pro Non-Diagnostické Prostředky In Vitro
určit moje zdravotnických zařízení zapadá do „invazivní“ kategorii, zúžení mé hledání se stanoví Pravidla 1, 2 a 3.
po přezkoumání možností určím, že platí pravidlo 1.
na základě mého zamýšleného použití je můj zdravotnický prostředek považován za třídu II v Kanadě.,
URČENÍ VAŠÍ CESTY NA TRH V KANADĚ
k Dispozici jsou čtyři úrovně zdravotnických zařízení klasifikace v Kanadě:
- Třída
- II. Třídy
- III. Třídy
- IV. Třída
Před odchodem na trh v Kanadě, musíte nejprve požádat o lékařské zařízení licenci. Zdravotnické prostředky třídy I nevyžadují licenci. Výrobci mohou odkazovat na Health Canada guidance document, který vás provede tímto procesem.,
Výrobci zdravotnických prostředků třídy III a třídy IV mohou získat licenci předložením aplikace premarket ve formátech ToC nebo Health Canada pro vstup na kanadský trh.
budete také muset získat certifikaci ISO 13485 s mdsap.
Aktualizace Health Canada Předpisy: MDSAP
od ledna 2019, všechny lékařské výrobci zařízení prodávat zdravotnické prostředky Třídy II a vyšší na Kanadský trh, musí být součástí Zdravotnického Prostředku, Jednotného Auditu Programu (MDSAP).,
tito výrobci musí projít a projít úplným auditem svého systému řízení kvality (QMS) prostřednictvím programu v Kanadě. V současné době existuje 6 regionů po celém světě, které se účastní MDSAP, včetně Kanady, USA, Japonska, Brazílie a Austrálie.
kromě těch výrobců, které společnost Health Canada požaduje k účasti na programu, je účast v MDSAP pro výrobce volitelná.,
získat MDSAP certifikace, lékařské výrobci zařízení musí splnit následující tři kroky:
- Aplikace a hodnocení
- Off-site dokumentaci auditu
- On-site audit
Zařízení výrobci prodávají do Kanady bude předmětem každoročního hodnocení, s recertifikační audit každý třetí rok. Soulad s programem jednotného auditu zdravotnických prostředků je založen na splnění pokynů normy ISO 13485 o systémech řízení kvality zdravotnických prostředků.,
pro zefektivnění aplikace doporučujeme proškolení vašich produktových a kvalitativních a regulačních týmů v příslušných požadavcích MDSAP.
Naše bezplatné posouzení nedostatků nástroj pro MDSAP a ISO 13485 pomáhá výrobcům zařízení posoudit QMS pokynů z normy ISO vedle požadavky Health Canada audit organizace (AO) a dalších regionů, které se účastní programu.,
závěr
nyní máte informace a zdroje, které potřebujete k určení váš zdravotní klasifikace zařízení a cestu na trh ve třech z největších tržišť na celém světě.
bez ohledu na klasifikaci vašeho zařízení je nutné dodržovat regulační pokyny, které platí na každém trhu. Splnění shody je klíčovým aspektem řízení kvality, který nakonec rozhodne o osudu vašeho zařízení a společnosti jako celku.,
na Greenlight Guru si vážíme důležitosti řízení kvality zdravotnických prostředků (MDQMS). Náš software MDQMS je jediným řešením na světě navrženým speciálně pro zdravotnické prostředky. Je v souladu s nejnovějšími průmyslovými standardními osvědčenými postupy pro správu kontrol a rizik návrhu produktu, jakož i kontrolních činností a dalších událostí kvality, které poskytují plnou sledovatelnost po celou dobu životnosti vašeho zdravotnického zařízení.
hledáte řešení pro řízení designu, které vám pomůže přinést bezpečnější zdravotnické prostředky na trh rychleji s menším rizikem?, Klikněte zde pro rychlou prohlídku zdravotnického zařízení Greenlight Guru QMS software →